Thompson Bryony A, Walters Rhiannon, Parsons Michael T, Dumenil Troy, Drost Mark, Tiersma Yvonne, Lindor Noralane M, Tavtigian Sean V, de Wind Niels, Spurdle Amanda B
Department of Pathology, The Royal Melbourne Hospital, Melbourne, VIC, Australia.
Department of Clinical Pathology, The University of Melbourne, Melbourne, VIC, Australia.
Front Genet. 2020 Jul 27;11:798. doi: 10.3389/fgene.2020.00798. eCollection 2020.
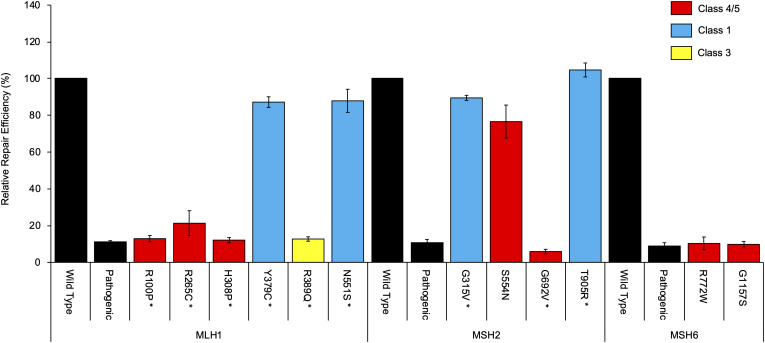
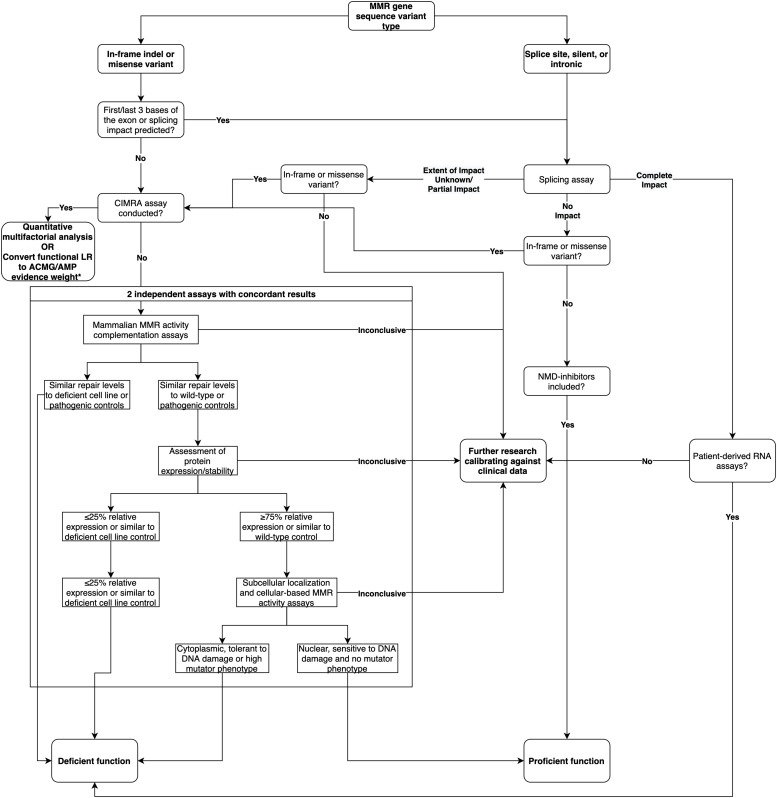
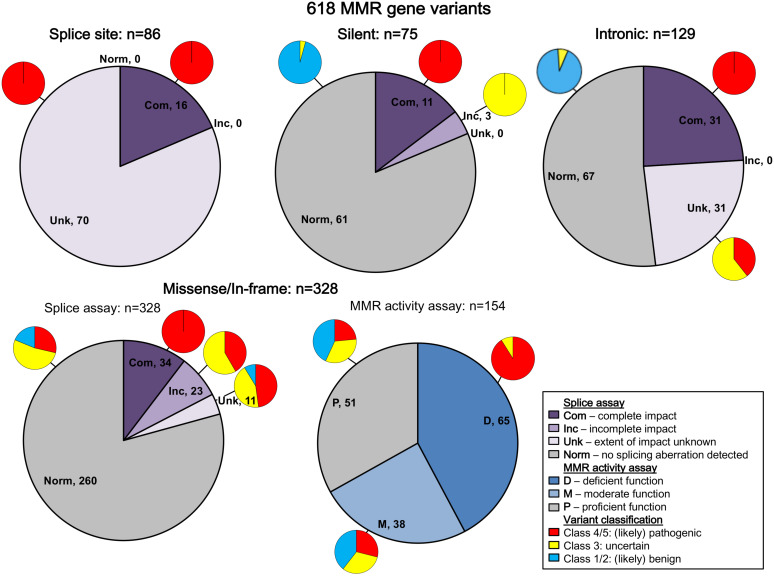
Functional assays that assess mRNA splicing can be used in interpretation of the clinical significance of sequence variants, including the Lynch syndrome-associated mismatch repair (MMR) genes. The purpose of this study was to investigate the contribution of splicing assay data to the classification of MMR gene sequence variants. We assayed mRNA splicing for 24 sequence variants in , , and , including 12 missense variants that were also assessed using a cell-free MMR activity (CIMRA) assay. Multifactorial likelihood analysis was conducted for each variant, combining CIMRA outputs and clinical data where available. We collated these results with existing public data to provide a dataset of splicing assay results for a total of 671 MMR gene sequence variants (328 missense/in-frame indel), and published and unpublished repair activity measurements for 154 of these variants. There were 241 variants for which a splicing aberration was detected: 92 complete impact, 33 incomplete impact, and 116 where it was not possible to determine complete versus incomplete splicing impact. Splicing results mostly aided in the interpretation of intronic (72%) and silent (92%) variants and were the least useful for missense substitutions/in-frame indels (10%). MMR protein functional activity assays were more useful in the analysis of these exonic variants but by design they were not able to detect clinically important splicing aberrations identified by parallel mRNA assays. The development of high throughput assays that can quantitatively assess impact on mRNA transcript expression and protein function in parallel will streamline classification of MMR gene sequence variants.
评估mRNA剪接的功能分析可用于解释序列变异的临床意义,包括与林奇综合征相关的错配修复(MMR)基因。本研究的目的是调查剪接分析数据对MMR基因序列变异分类的贡献。我们对MLH1、MSH2和MSH6中的24个序列变异进行了mRNA剪接检测,其中包括12个错义变异,这些变异也使用无细胞MMR活性(CIMRA)分析进行了评估。对每个变异进行多因素似然分析,将CIMRA结果与可用的临床数据相结合。我们将这些结果与现有的公共数据进行整理,以提供一个包含671个MMR基因序列变异(328个错义/框内插入缺失)的剪接分析结果数据集,以及其中154个变异已发表和未发表的修复活性测量数据。有241个变异检测到剪接异常:92个完全影响,33个不完全影响,116个无法确定完全与不完全剪接影响。剪接结果大多有助于内含子变异(72%)和沉默变异(92%)的解释,对错义替代/框内插入缺失变异的解释作用最小(10%)。MMR蛋白功能活性分析在这些外显子变异的分析中更有用,但根据设计,它们无法检测到平行mRNA分析鉴定的临床重要剪接异常。能够并行定量评估对mRNA转录表达和蛋白质功能影响的高通量分析的开发将简化MMR基因序列变异的分类。