Institute for Frontier Life and Medical Sciences, Kyoto University, 53 Shougoin Kawara-cho, Sakyo-ku, Kyoto, 606-8507, Japan.
Institute for Liberal Arts and Sciences, Kyoto University, Yoshidanihonmatsu-cho, Sakyo-ku, Kyoto, 606-8501, Japan.
Nat Commun. 2020 Aug 28;11(1):4318. doi: 10.1038/s41467-020-17900-3.
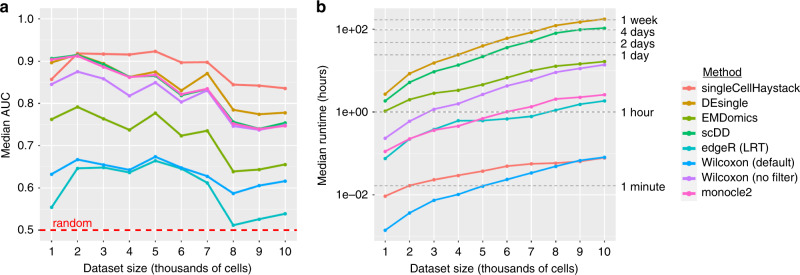
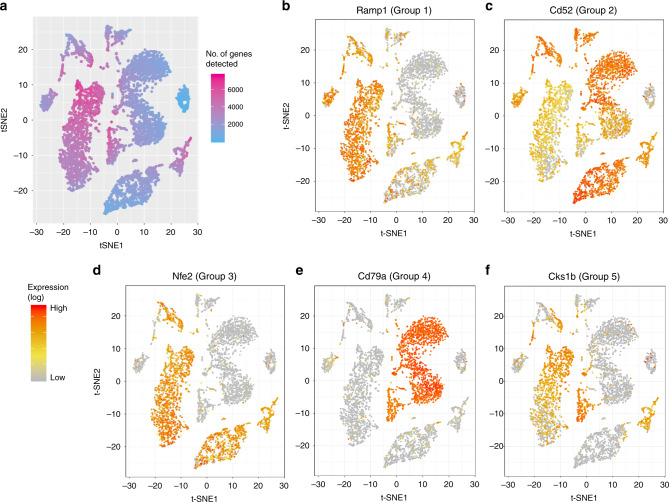
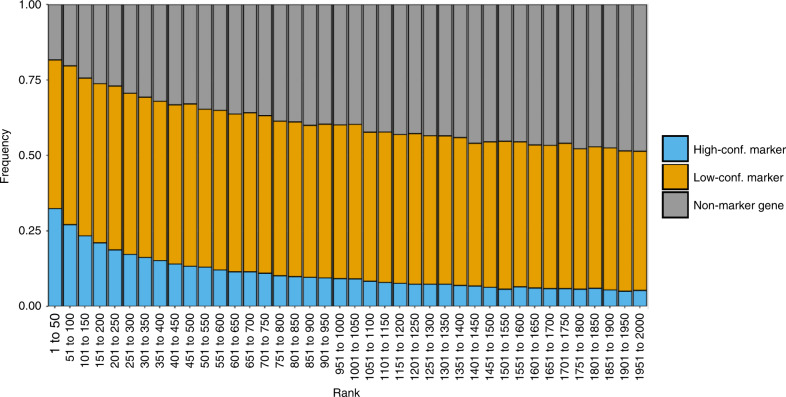
A common analysis of single-cell sequencing data includes clustering of cells and identifying differentially expressed genes (DEGs). How cell clusters are defined has important consequences for downstream analyses and the interpretation of results, but is often not straightforward. To address this difficulty, we present singleCellHaystack, a method that enables the prediction of DEGs without relying on explicit clustering of cells. Our method uses Kullback-Leibler divergence to find genes that are expressed in subsets of cells that are non-randomly positioned in a multidimensional space. Comparisons with existing DEG prediction approaches on artificial datasets show that singleCellHaystack has higher accuracy. We illustrate the usage of singleCellHaystack through applications on 136 real transcriptome datasets and a spatial transcriptomics dataset. We demonstrate that our method is a fast and accurate approach for DEG prediction in single-cell data. singleCellHaystack is implemented as an R package and is available from CRAN and GitHub.
单细胞测序数据分析通常包括细胞聚类和差异表达基因(DEGs)的识别。细胞聚类的定义对下游分析和结果解释有重要影响,但通常并不简单。为了解决这个难题,我们提出了 singleCellHaystack 方法,该方法可以在不依赖细胞显式聚类的情况下预测 DEGs。我们的方法使用 Kullback-Leibler 散度来找到在多维空间中随机定位的细胞亚群中表达的基因。在人工数据集上与现有 DEG 预测方法的比较表明,singleCellHaystack 具有更高的准确性。我们通过对 136 个真实转录组数据集和一个空间转录组数据集的应用说明了 singleCellHaystack 的用法。我们证明了我们的方法是单细胞数据中 DEG 预测的一种快速而准确的方法。singleCellHaystack 作为一个 R 包实现,可从 CRAN 和 GitHub 获得。