Pathogen and Microbiome Institute, Northern Arizona University, Flagstaff, Arizona, USA.
Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, Arizona, USA.
mBio. 2020 Sep 4;11(5):e02107-20. doi: 10.1128/mBio.02107-20.
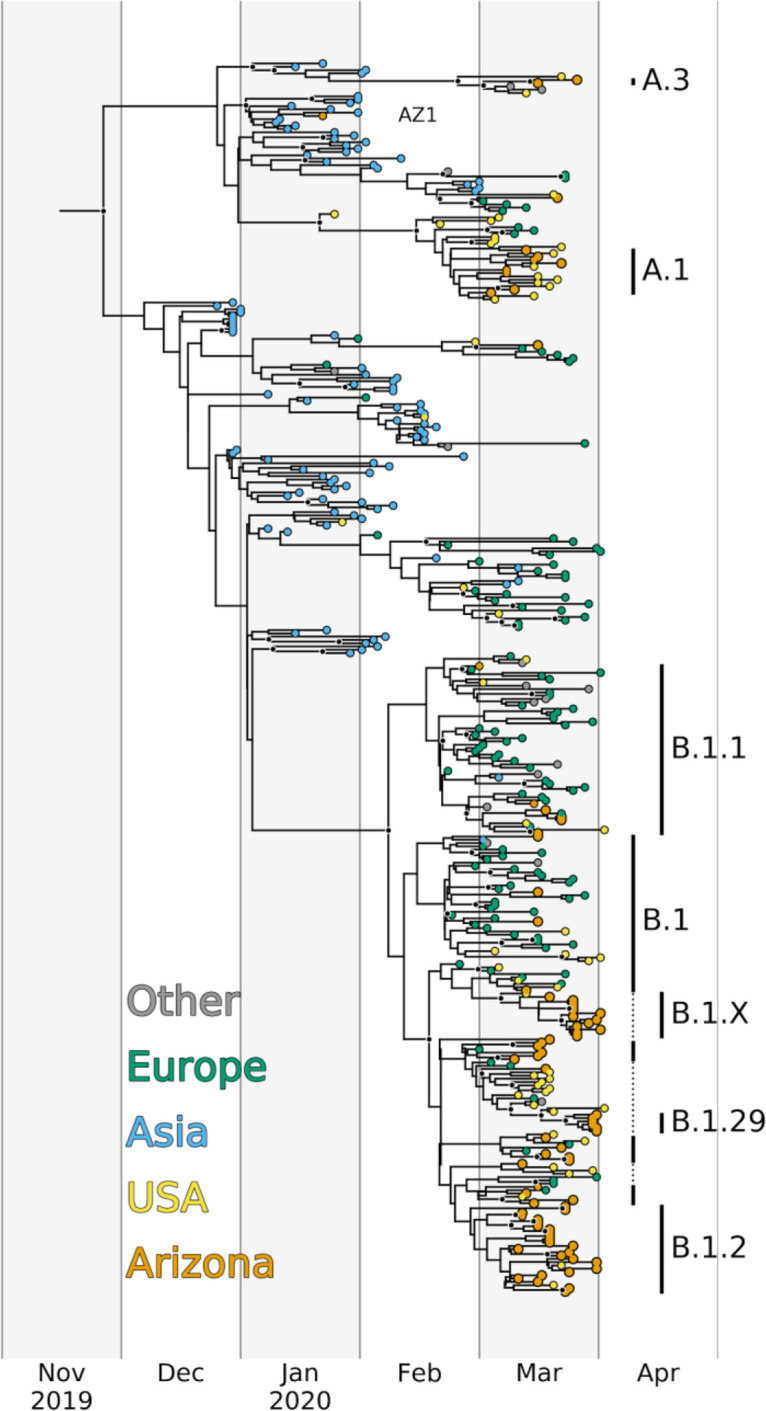
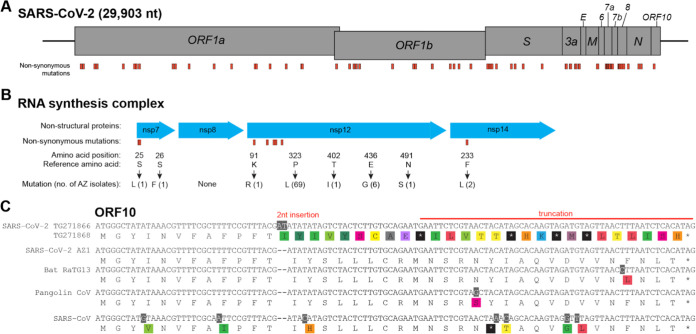
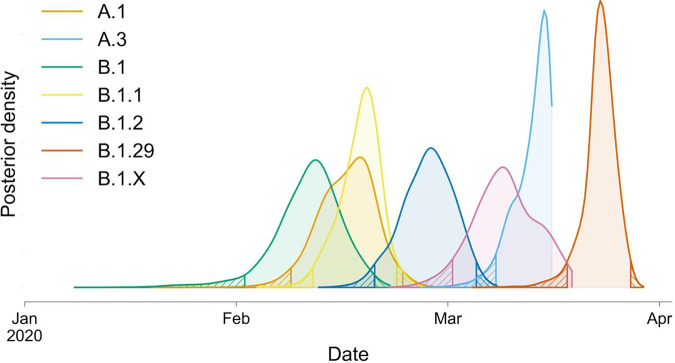
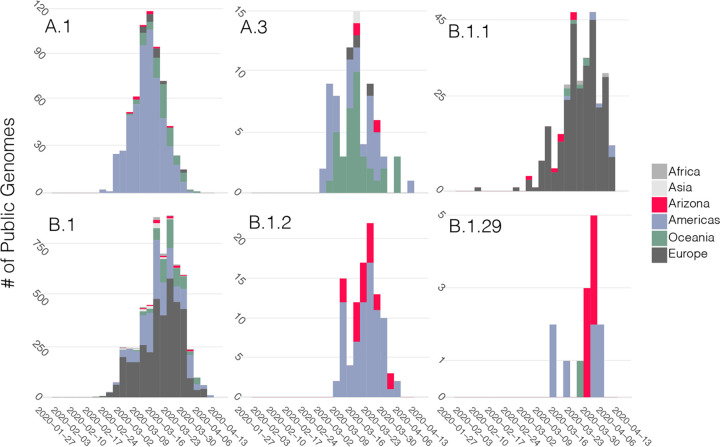
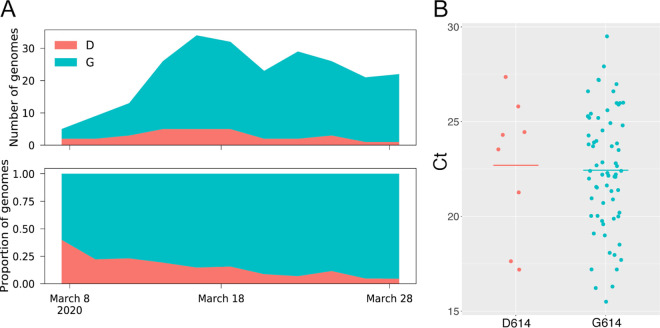
In December of 2019, a novel coronavirus, SARS-CoV-2, emerged in the city of Wuhan, China, causing severe morbidity and mortality. Since then, the virus has swept across the globe, causing millions of confirmed infections and hundreds of thousands of deaths. To better understand the nature of the pandemic and the introduction and spread of the virus in Arizona, we sequenced viral genomes from clinical samples tested at the TGen North Clinical Laboratory, the Arizona Department of Health Services, and those collected as part of community surveillance projects at Arizona State University and the University of Arizona. Phylogenetic analysis of 84 genomes from across Arizona revealed a minimum of 11 distinct introductions inferred to have occurred during February and March. We show that >80% of our sequences descend from strains that were initially circulating widely in Europe but have since dominated the outbreak in the United States. In addition, we show that the first reported case of community transmission in Arizona descended from the Washington state outbreak that was discovered in late February. Notably, none of the observed transmission clusters are epidemiologically linked to the original travel-related case in the state, suggesting successful early isolation and quarantine. Finally, we use molecular clock analyses to demonstrate a lack of identifiable, widespread cryptic transmission in Arizona prior to the middle of February 2020. As the COVID-19 pandemic swept across the United States, there was great differential impact on local and regional communities. One of the earliest and hardest hit regions was in New York, while at the same time Arizona (for example) had low incidence. That situation has changed dramatically, with Arizona now having the highest rate of disease increase in the country. Understanding the roots of the pandemic during the initial months is essential as the pandemic continues and reaches new heights. Genomic analysis and phylogenetic modeling of SARS-COV-2 in Arizona can help to reconstruct population composition and predict the earliest undetected introductions. This foundational work represents the basis for future analysis and understanding as the pandemic continues.
2019 年 12 月,一种新型冠状病毒 SARS-CoV-2 在中国武汉市出现,导致严重的发病率和死亡率。此后,该病毒席卷全球,导致数百万人感染和数十万人死亡。为了更好地了解疫情的性质以及病毒在亚利桑那州的传入和传播,我们对 TGen North 临床实验室、亚利桑那州卫生署以及亚利桑那州立大学和亚利桑那大学社区监测项目中收集的临床样本进行了病毒基因组测序。对来自亚利桑那州的 84 个基因组的系统发育分析显示,至少有 11 个不同的传入事件发生在 2 月和 3 月期间。我们表明,我们的序列中超过 80%来自最初在欧洲广泛传播但此后在美国疫情中占主导地位的菌株。此外,我们还表明,亚利桑那州首例社区传播病例来自 2 月底发现的华盛顿州疫情。值得注意的是,观察到的传播群没有一个与该州最初的与旅行相关的病例在流行病学上有联系,这表明成功地进行了早期隔离和检疫。最后,我们使用分子钟分析表明,在 2020 年 2 月中旬之前,亚利桑那州没有可识别的广泛隐性传播。随着 COVID-19 疫情席卷美国,对当地和地区社区造成了巨大的不同影响。最早和受打击最严重的地区之一是纽约,而与此同时亚利桑那州(例如)发病率较低。这种情况发生了巨大变化,亚利桑那州现在是美国疾病发病率最高的州。随着疫情的持续和达到新的高度,了解疫情初期的情况至关重要。对亚利桑那州 SARS-COV-2 的基因组分析和系统发育建模可以帮助重建人群构成并预测最早的未检测到的传入事件。这项基础工作代表了疫情持续期间未来分析和理解的基础。