Pathogen Genomics Center, National Institute of Infectious Diseases, Tokyo, Japan.
Sapporo City Institute of Public Health, Sapporo, Japan.
mSphere. 2020 Nov 11;5(6):e00786-20. doi: 10.1128/mSphere.00786-20.
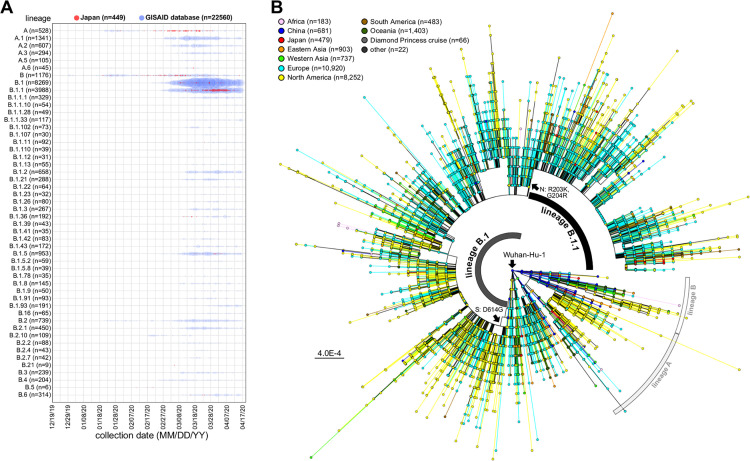
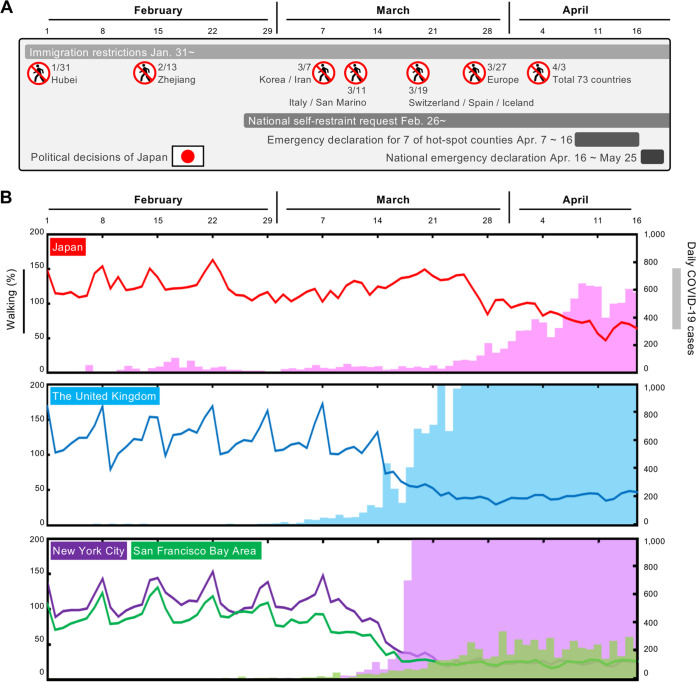
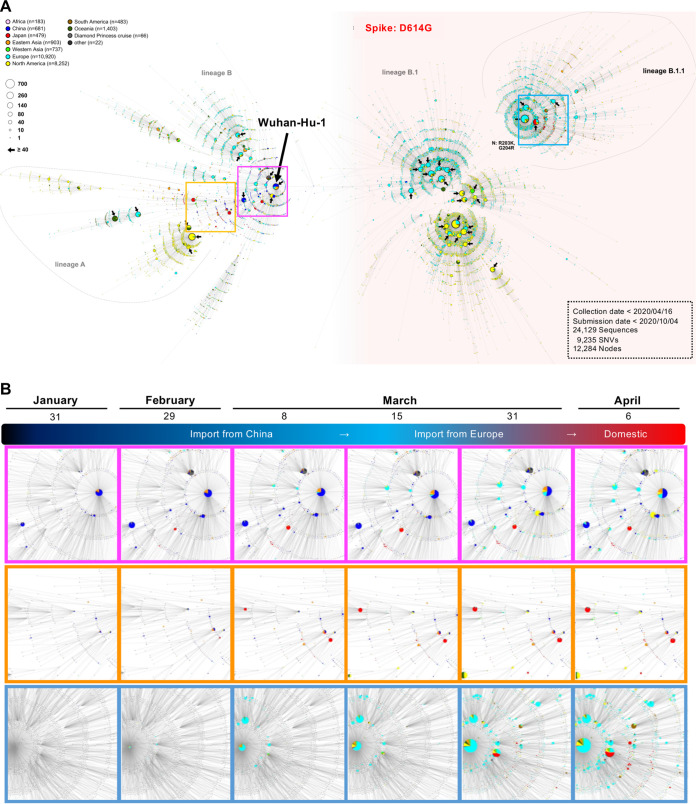
After the first case of coronavirus disease 2019 (COVID-19) in Japan on 15 January 2020, multiple nationwide COVID-19 clusters were identified by the end of February. The Japanese government focused on mitigating the emerging COVID-19 clusters by conducting active nationwide epidemiological surveillance. However, an increasing number of cases continued to appear until early April 2020, many with unclear infection routes and no recent history of travel outside Japan. We aimed to evaluate the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome sequences from the COVID-19 cases that appeared until early April 2020 and to characterize their genealogical networks in order to demonstrate possible routes of spread in Japan. Nasopharyngeal specimens were collected from patients, and reverse transcription-quantitative PCR tests for SARS-CoV-2 were performed. Positive RNA samples were subjected to whole-genome sequencing, and a haplotype network analysis was performed. Some of the primary clusters identified during January and February 2020 in Japan descended directly from the Wuhan-Hu-1-related isolates from China and other distinct clusters. Clusters were almost contained until mid-March; the haplotype network analysis demonstrated that the COVID-19 cases from late March through early April may have created an additional large cluster related to the outbreak in Europe, leading to additional spread within Japan. In conclusion, genome surveillance has suggested that there were at least two distinct SARS-CoV-2 introductions into Japan from China and other countries. This study aimed to evaluate the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome sequences from COVID-19 cases and to characterize their genealogical networks to demonstrate possible routes of spread in Japan. We found that there were at least two distinct SARS-CoV-2 introductions into Japan, initially from China and subsequently from other countries, including Europe. Our findings can help understand how SARS-CoV-2 entered Japan and contribute to increased knowledge of SARS-CoV-2 in Asia and its association with implemented stay-at-home/shelter-in-place/self-restraint/lockdown measures. This study suggested that it is necessary to formulate a more efficient containment strategy using real-time genome surveillance to support epidemiological field investigations in order to highlight potential infection linkages and mitigate the next wave of COVID-19 in Japan.
自 2020 年 1 月 15 日日本首例 2019 年冠状病毒病(COVID-19)病例以来,2 月底已确定多个全国性 COVID-19 集群。日本政府通过开展积极的全国性流行病学监测,专注于减轻新出现的 COVID-19 集群。然而,病例数量持续增加,直到 2020 年 4 月初,许多病例的感染途径不明确,且近期无日本境外旅行史。我们旨在评估截至 2020 年 4 月初出现的 COVID-19 病例的严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)基因组序列,并对其系统发育网络进行特征描述,以展示日本可能的传播途径。我们从患者中采集鼻咽标本,进行 SARS-CoV-2 的逆转录定量 PCR 检测。对阳性 RNA 样本进行全基因组测序,并进行单倍型网络分析。2020 年 1 月和 2 月期间确定的一些主要集群直接源自中国武汉-Hu-1 相关分离株和其他不同的集群。集群几乎在 3 月中旬前被包含在内;单倍型网络分析表明,3 月底至 4 月初的 COVID-19 病例可能形成了一个与欧洲暴发相关的额外大集群,从而导致日本内部的进一步传播。总之,基因组监测表明,至少有两批 SARS-CoV-2 从中国和其他国家传入日本。本研究旨在评估 COVID-19 病例的严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)基因组序列,并对其系统发育网络进行特征描述,以展示日本可能的传播途径。我们发现,至少有两批 SARS-CoV-2 从中国和其他国家传入日本,最初源自中国,随后源自包括欧洲在内的其他国家。我们的研究结果可以帮助了解 SARS-CoV-2 如何进入日本,并有助于增加对亚洲 SARS-CoV-2 的了解及其与实施的居家隔离/就地避难/自我约束/封锁措施的关联。本研究表明,有必要利用实时基因组监测制定更有效的遏制策略,以支持流行病学现场调查,从而突出潜在的感染联系,并减轻日本下一波 COVID-19 的影响。