Department of Pediatrics, Child Neurology Unit, Presidio Ospedaliero Provinciale Santa Maria Nuova Azienda USL-IRCCS di Reggio Emilia, Reggio Emilia, Italy.
Child Neuropsychiatric Unit, Department of Medicine and Surgery, University of Parma, Parma, Italy.
Acta Biomed. 2020 Sep 7;91(3):e2020075. doi: 10.23750/abm.v91i3.9272.
Niemann-Pick disease type C (NPC) is a lysosomal storage disease caused by mutations in NPC1 or NPC2 genes.
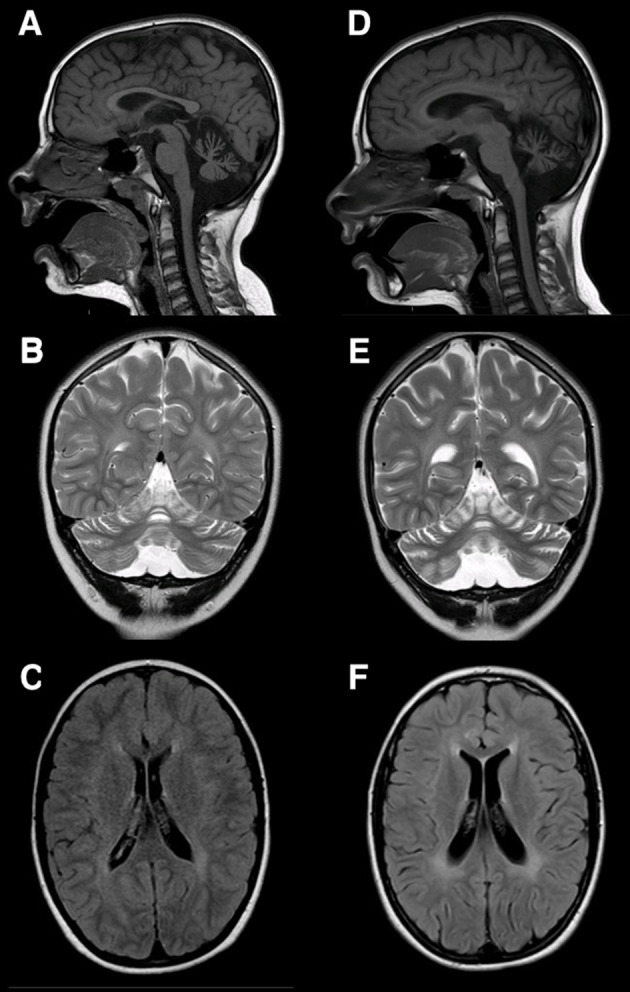
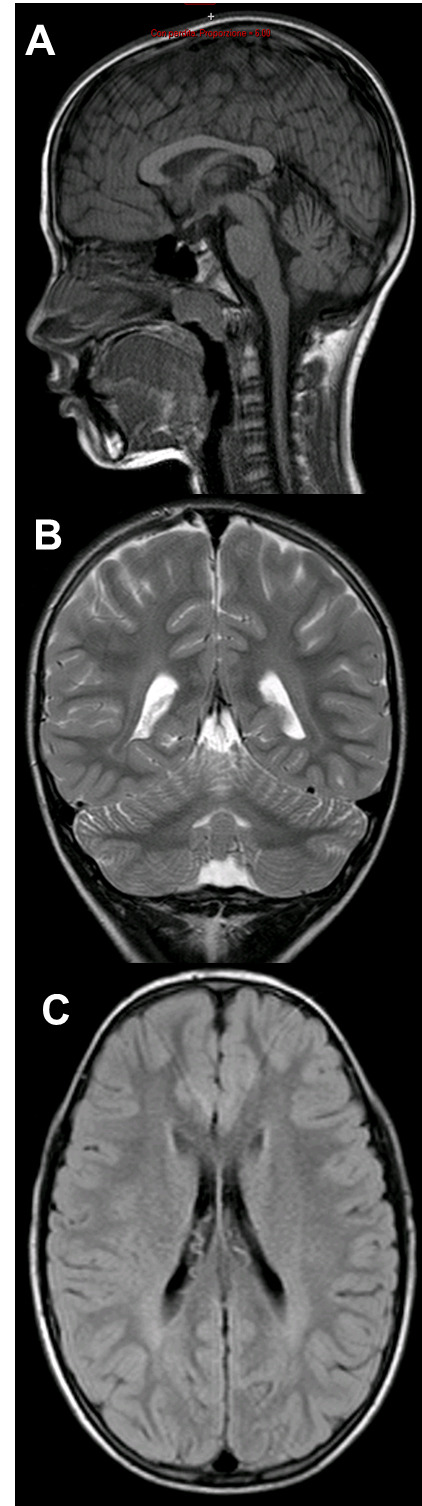
We present two brothers with the same compound heterozygous variants in exon 13 of the NPC1 gene (18q11.2), the first one (c.1955C> G, p. Ser652Trp), inherited from the mother, the second (c.2107T>A p.Phe703Ile) inherited from the father, associated to the classical biochemical phenotype of NPC. The two brothers presented unspecific neurologic symptoms with difference in age of onset: one presented dyspraxia and motor clumsiness at age 7 years, the other showed a systemic presentation with hepatosplenomegaly noted at the age of two months and neurological symptoms onset at age 4 with speech disturbance. Clinical evolution and neuroimaging data led to the final diagnosis. Systemic signs did not correlate with the onset of neurological symptoms. Miglustat therapy was started in both patients.
We highlight the extreme phenotypic heterogeneity of NP-C in the presence of the same genetic variant and the unspecificity of neurologic signs at onset as previously reported. We report some positive effects of miglustat on disease progression assessed also with neuropsychological follow-up, with an age-dependent response.
尼曼-匹克病 C 型(NPC)是一种溶酶体贮积病,由 NPC1 或 NPC2 基因突变引起。
我们介绍了两位兄弟,他们在 NPC1 基因的exon 13 中携带相同的复合杂合变异(18q11.2),第一个变异(c.1955C>G,p.Ser652Trp)来自母亲,第二个变异(c.2107T>A,p.Phe703Ile)来自父亲,与 NPC 的经典生化表型相关。这两兄弟表现出非特异性的神经症状,但发病年龄不同:一个在 7 岁时出现运动协调障碍和运动笨拙,另一个在 2 个月时出现肝脾肿大,4 岁时出现神经症状,表现为言语障碍。临床演变和神经影像学数据导致了最终诊断。全身症状与神经症状的出现无关。两兄弟均开始接受米格列醇治疗。
我们强调了 NP-C 在存在相同遗传变异的情况下存在极端表型异质性,以及神经症状在发病时的非特异性,这与之前的报道一致。我们报告了米格列醇治疗对疾病进展的一些积极影响,也通过神经心理学随访进行了评估,其疗效具有年龄依赖性。