Institut National de la Santé et de la Recherche Médicale, Unité 820, Faculté de Médecine Lyon-Est Claude Bernard, 7 Rue G, Paradin, F-69008, Lyon, France.
Orphanet J Rare Dis. 2010 Jun 3;5:16. doi: 10.1186/1750-1172-5-16.
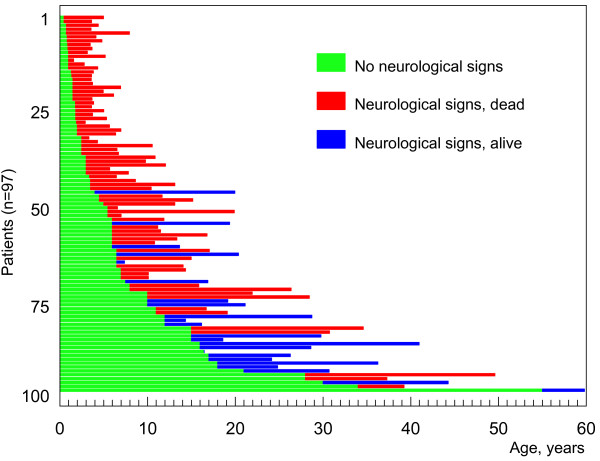
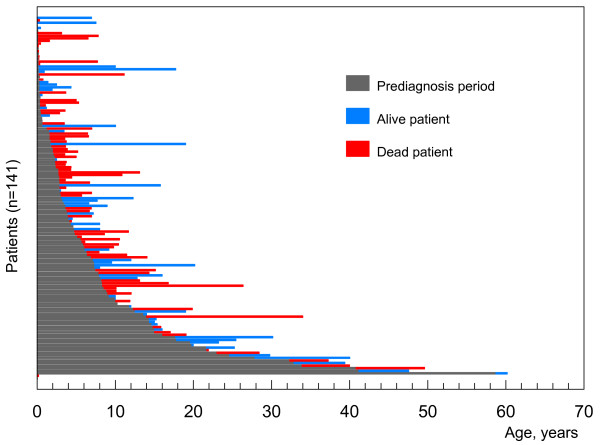
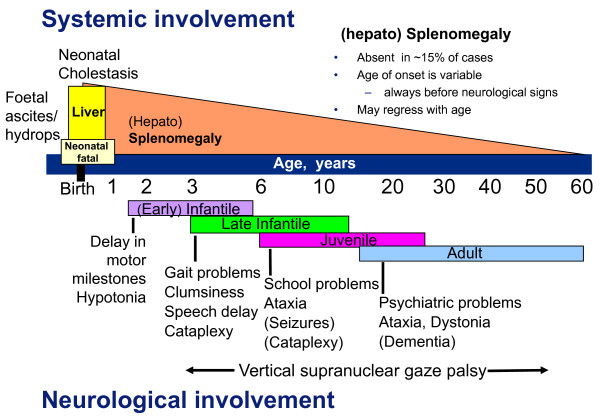
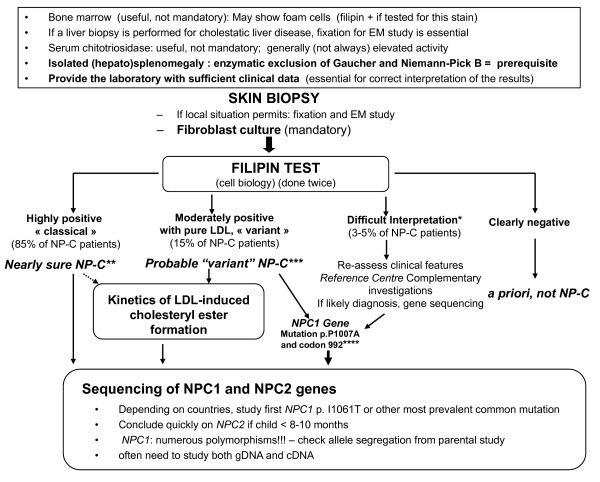
Niemann-Pick C disease (NP-C) is a neurovisceral atypical lysosomal lipid storage disorder with an estimated minimal incidence of 1/120,000 live births. The broad clinical spectrum ranges from a neonatal rapidly fatal disorder to an adult-onset chronic neurodegenerative disease. The neurological involvement defines the disease severity in most patients but is typically preceded by systemic signs (cholestatic jaundice in the neonatal period or isolated spleno- or hepatosplenomegaly in infancy or childhood). The first neurological symptoms vary with age of onset: delay in developmental motor milestones (early infantile period), gait problems, falls, clumsiness, cataplexy, school problems (late infantile and juvenile period), and ataxia not unfrequently following initial psychiatric disturbances (adult form). The most characteristic sign is vertical supranuclear gaze palsy. The neurological disorder consists mainly of cerebellar ataxia, dysarthria, dysphagia, and progressive dementia. Cataplexy, seizures and dystonia are other common features. NP-C is transmitted in an autosomal recessive manner and is caused by mutations of either the NPC1 (95% of families) or the NPC2 genes. The exact functions of the NPC1 and NPC2 proteins are still unclear. NP-C is currently described as a cellular cholesterol trafficking defect but in the brain, the prominently stored lipids are gangliosides. Clinical examination should include comprehensive neurological and ophthalmological evaluations. The primary laboratory diagnosis requires living skin fibroblasts to demonstrate accumulation of unesterified cholesterol in perinuclear vesicles (lysosomes) after staining with filipin. Pronounced abnormalities are observed in about 80% of the cases, mild to moderate alterations in the remainder ("variant" biochemical phenotype). Genotyping of patients is useful to confirm the diagnosis in the latter patients and essential for future prenatal diagnosis. The differential diagnosis may include other lipidoses; idiopathic neonatal hepatitis and other causes of cholestatic icterus should be considered in neonates, and conditions with cerebellar ataxia, dystonia, cataplexy and supranuclear gaze palsy in older children and adults. Symptomatic management of patients is crucial. A first product, miglustat, has been granted marketing authorization in Europe and several other countries for specific treatment of the neurological manifestations. The prognosis largely correlates with the age at onset of the neurological manifestations.
尼曼-皮克 C 病(NP-C)是一种神经内脏非典型溶酶体脂质贮积症,估计活产儿的最低发病率为 1/120000。广泛的临床谱范围从新生儿迅速致命的疾病到成人发病的慢性神经退行性疾病。神经系统受累定义了大多数患者的疾病严重程度,但通常先于全身症状(新生儿期胆汁淤积性黄疸或婴儿期或儿童期孤立性脾肿大或肝脾肿大)。首发的神经系统症状随发病年龄而变化:发育运动里程碑延迟(婴儿早期)、步态问题、跌倒、笨拙、猝倒、学习问题(婴儿晚期和青少年期),以及初始精神障碍后常出现的共济失调(成人型)。最特征性的体征是垂直性核上性眼球运动障碍。神经系统疾病主要由小脑共济失调、构音障碍、吞咽困难和进行性痴呆组成。猝倒、癫痫发作和肌张力障碍也是常见特征。NP-C 以常染色体隐性方式遗传,由 NPC1(95%的家庭)或 NPC2 基因突变引起。NPC1 和 NPC2 蛋白的确切功能仍不清楚。NP-C 目前被描述为一种细胞胆固醇转运缺陷,但在大脑中,储存的主要脂质是神经节苷脂。临床检查应包括全面的神经学和眼科评估。初步实验室诊断需要活皮肤成纤维细胞,在使用 filipin 染色后,证明未酯化胆固醇在核周小泡(溶酶体)中的积累。大约 80%的病例观察到明显异常,其余病例为轻度至中度改变(“变异”生化表型)。对患者进行基因分型有助于在后者中确认诊断,并对未来的产前诊断至关重要。鉴别诊断可能包括其他脂质贮积症;新生儿期应考虑特发性新生儿肝炎和其他胆汁淤积性黄疸的原因,而在较大儿童和成人中,应考虑小脑共济失调、肌张力障碍、猝倒和核上性眼球运动障碍的疾病。患者的对症治疗至关重要。第一个产品米格列醇已在欧洲和其他几个国家获得上市许可,用于治疗特定的神经表现。预后与神经表现的发病年龄密切相关。