Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA; Stanford Neurosciences Interdepartmental Program, Stanford University School of Medicine, Stanford, CA 94305, USA.
Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA; Department of Biology, Stanford University, Stanford, CA 94305, USA.
Neuron. 2020 Dec 9;108(5):822-842. doi: 10.1016/j.neuron.2020.08.022. Epub 2020 Sep 14.
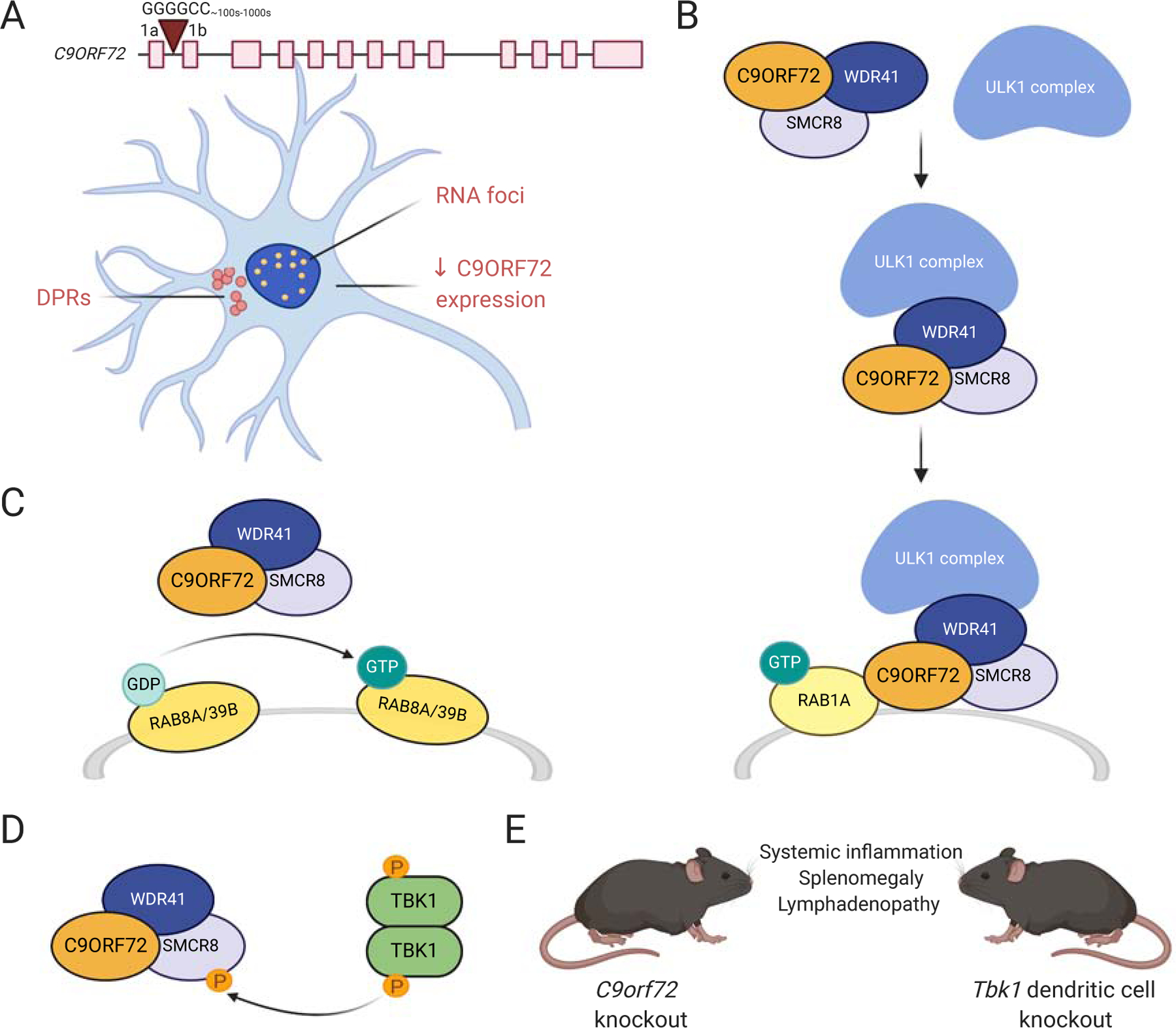
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder caused by the loss of motor neurons from the brain and spinal cord. The ALS community has made remarkable strides over three decades by identifying novel familial mutations, generating animal models, elucidating molecular mechanisms, and ultimately developing promising new therapeutic approaches. Some of these approaches reduce the expression of mutant genes and are in human clinical trials, highlighting the need to carefully consider the normal functions of these genes and potential contribution of gene loss-of-function to ALS. Here, we highlight known loss-of-function mechanisms underlying ALS, potential consequences of lowering levels of gene products, and the need to consider both gain and loss of function to develop safe and effective therapeutic strategies.
肌萎缩侧索硬化症(ALS)是一种致命的神经退行性疾病,由大脑和脊髓中的运动神经元丧失引起。在过去的三十年中,ALS 研究社区通过鉴定新的家族性突变、生成动物模型、阐明分子机制,最终开发出有前途的新治疗方法,取得了显著进展。其中一些方法可以降低突变基因的表达,并正在进行人体临床试验,这突出表明需要仔细考虑这些基因的正常功能以及基因功能丧失对 ALS 的潜在贡献。在这里,我们重点介绍 ALS 的已知功能丧失机制、降低基因产物水平的潜在后果,以及在开发安全有效的治疗策略时需要考虑功能获得和功能丧失这两方面。