Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, USA.
BMC Bioinformatics. 2020 Sep 15;21(1):406. doi: 10.1186/s12859-020-03720-1.
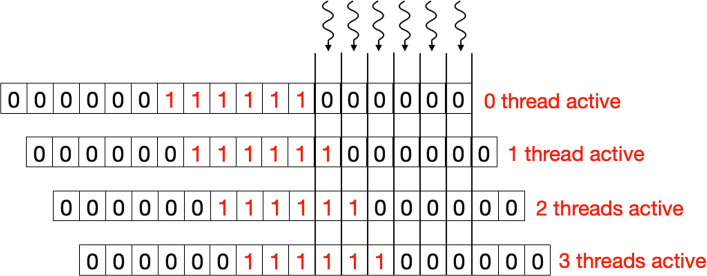
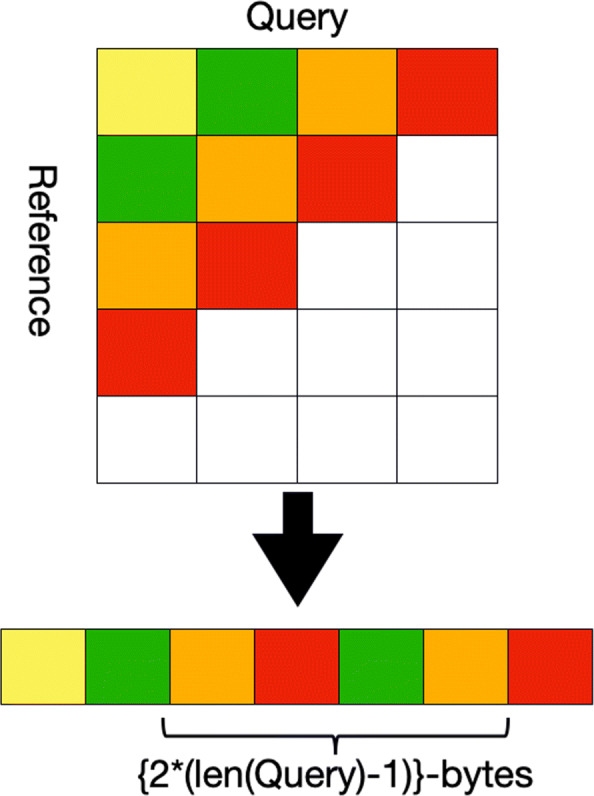
Bioinformatic workflows frequently make use of automated genome assembly and protein clustering tools. At the core of most of these tools, a significant portion of execution time is spent in determining optimal local alignment between two sequences. This task is performed with the Smith-Waterman algorithm, which is a dynamic programming based method. With the advent of modern sequencing technologies and increasing size of both genome and protein databases, a need for faster Smith-Waterman implementations has emerged. Multiple SIMD strategies for the Smith-Waterman algorithm are available for CPUs. However, with the move of HPC facilities towards accelerator based architectures, a need for an efficient GPU accelerated strategy has emerged. Existing GPU based strategies have either been optimized for a specific type of characters (Nucleotides or Amino Acids) or for only a handful of application use-cases.
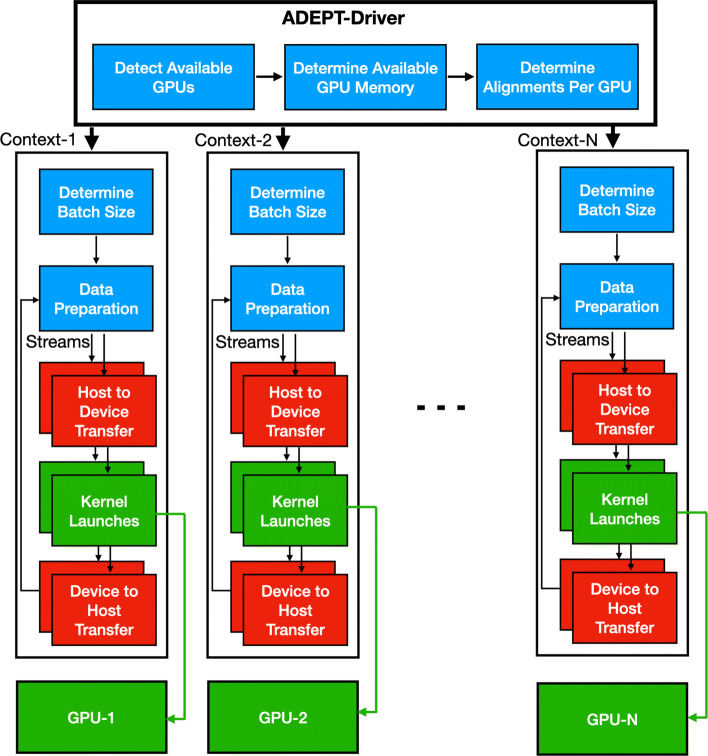
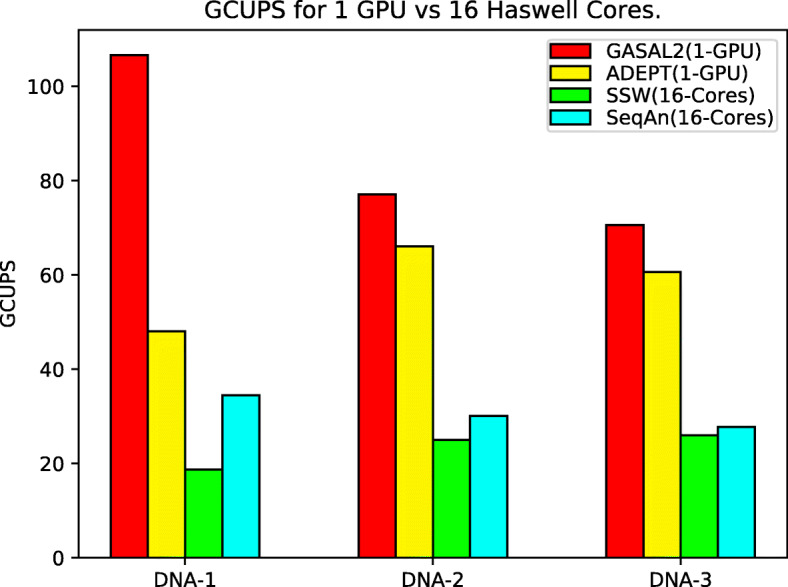
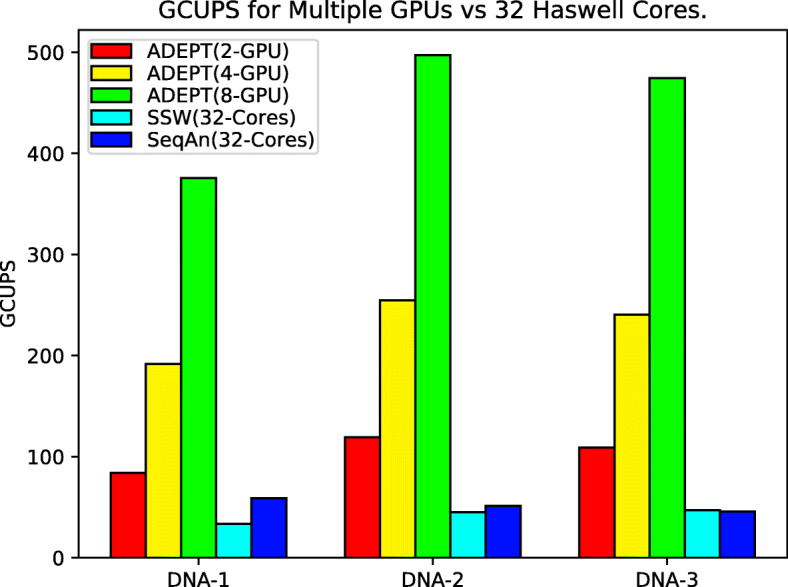
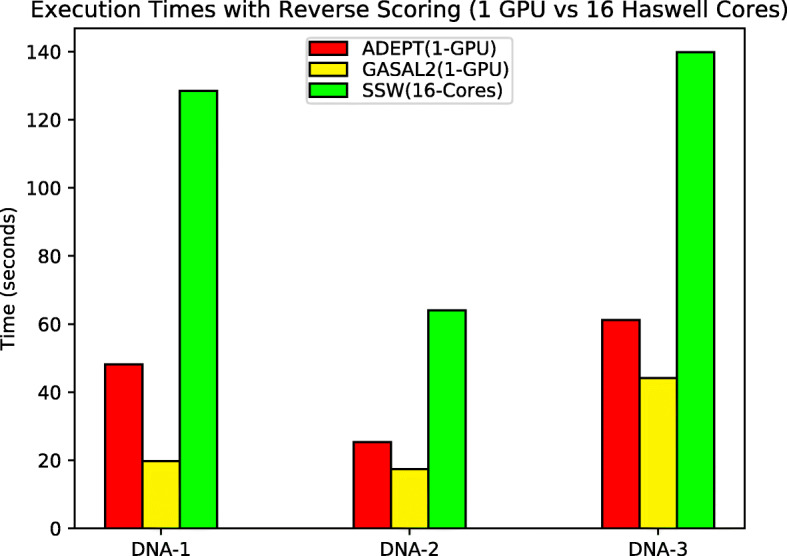
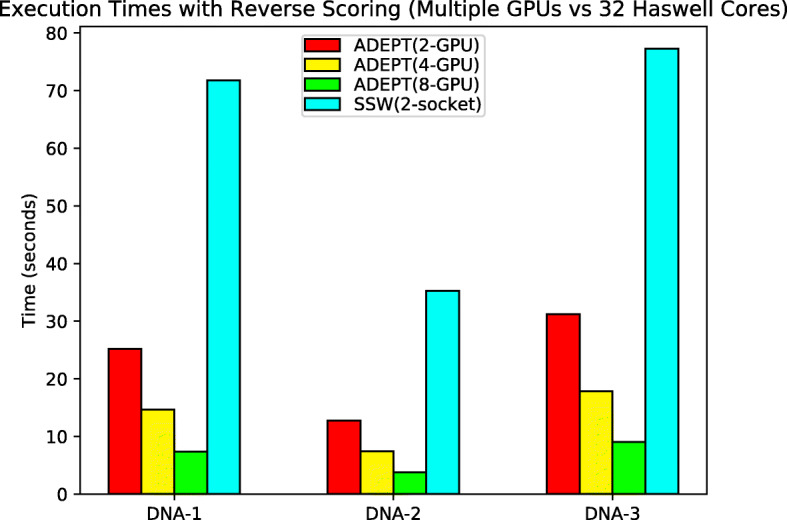
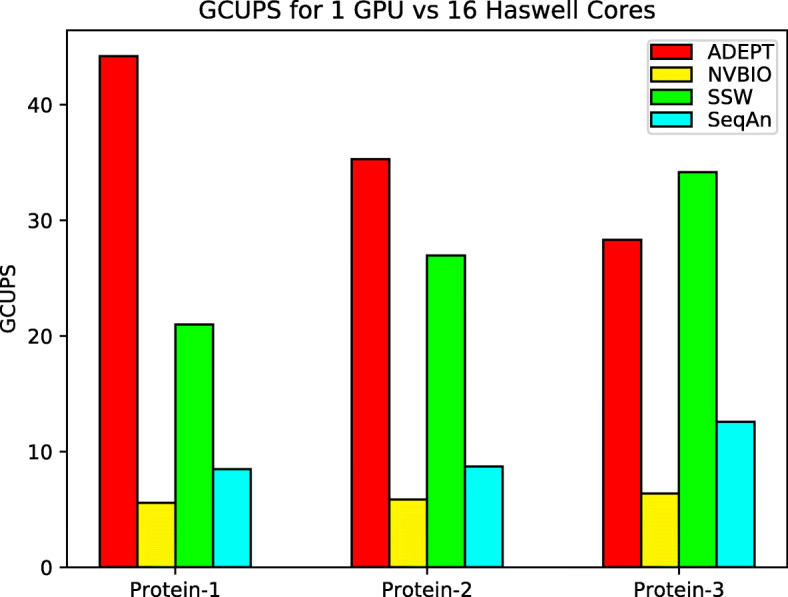
In this paper, we present ADEPT, a new sequence alignment strategy for GPU architectures that is domain independent, supporting alignment of sequences from both genomes and proteins. Our proposed strategy uses GPU specific optimizations that do not rely on the nature of sequence. We demonstrate the feasibility of this strategy by implementing the Smith-Waterman algorithm and comparing it to similar CPU strategies as well as the fastest known GPU methods for each domain. ADEPT's driver enables it to scale across multiple GPUs and allows easy integration into software pipelines which utilize large scale computational systems. We have shown that the ADEPT based Smith-Waterman algorithm demonstrates a peak performance of 360 GCUPS and 497 GCUPs for protein based and DNA based datasets respectively on a single GPU node (8 GPUs) of the Cori Supercomputer. Overall ADEPT shows 10x faster performance in a node-to-node comparison against a corresponding SIMD CPU implementation.
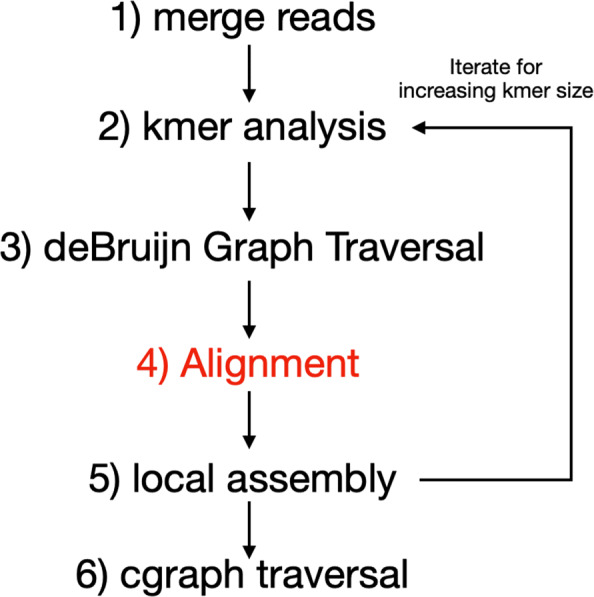
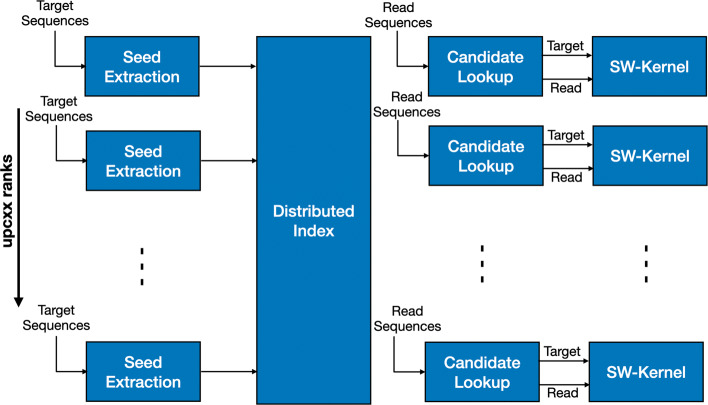
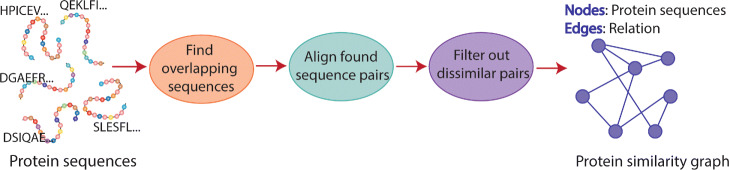
ADEPT demonstrates a performance that is either comparable or better than existing GPU strategies. We demonstrated the efficacy of ADEPT in supporting existing bionformatics software pipelines by integrating ADEPT in MetaHipMer a high-performance denovo metagenome assembler and PASTIS a high-performance protein similarity graph construction pipeline. Our results show 10% and 30% boost of performance in MetaHipMer and PASTIS respectively.
生物信息学工作流经常使用自动化基因组组装和蛋白质聚类工具。在这些工具的核心部分,大部分执行时间都花在确定两个序列之间的最佳局部比对上。这项任务是使用 Smith-Waterman 算法完成的,这是一种基于动态规划的方法。随着现代测序技术的出现和基因组与蛋白质数据库的不断增大,对更快的 Smith-Waterman 实现的需求也随之产生。CPU 有多种 SIMD 策略可用于 Smith-Waterman 算法。然而,随着 HPC 设施向基于加速器的架构转移,对高效 GPU 加速策略的需求也出现了。现有的基于 GPU 的策略要么针对特定类型的字符(核苷酸或氨基酸)进行了优化,要么仅针对少数应用用例进行了优化。
在本文中,我们提出了 ADEPT,这是一种针对 GPU 架构的新的序列对齐策略,它与领域无关,支持来自基因组和蛋白质的序列对齐。我们提出的策略使用了 GPU 特定的优化,不依赖于序列的性质。我们通过实现 Smith-Waterman 算法并将其与类似的 CPU 策略以及每个领域最快的已知 GPU 方法进行比较,证明了这种策略的可行性。ADEPT 的驱动程序使其能够在多个 GPU 上扩展,并允许轻松集成到利用大规模计算系统的软件管道中。我们已经表明,基于 ADEPT 的 Smith-Waterman 算法在单个 GPU 节点(Cori 超级计算机的 8 个 GPU)上分别针对基于蛋白质的和基于 DNA 的数据集实现了 360 GCUPS 和 497 GCUP 的峰值性能。总体而言,与相应的 SIMD CPU 实现相比,在节点到节点的比较中,ADEPT 的性能快了 10 倍。
ADEPT 的性能与现有 GPU 策略相当或更好。我们通过将 ADEPT 集成到高性能 de novo 宏基因组组装器 MetaHipMer 和高性能蛋白质相似性图构建管道 PASTIS 中,证明了 ADEPT 支持现有的生物信息学软件管道的有效性。我们的结果分别显示 MetaHipMer 和 PASTIS 的性能提升了 10%和 30%。