Department of Genetics and Genome Sciences, UCONN Health, 400 Farmington Avenue, Farmington, CT 06030, USA.
Open Biol. 2020 Sep;10(9):200195. doi: 10.1098/rsob.200195. Epub 2020 Sep 23.
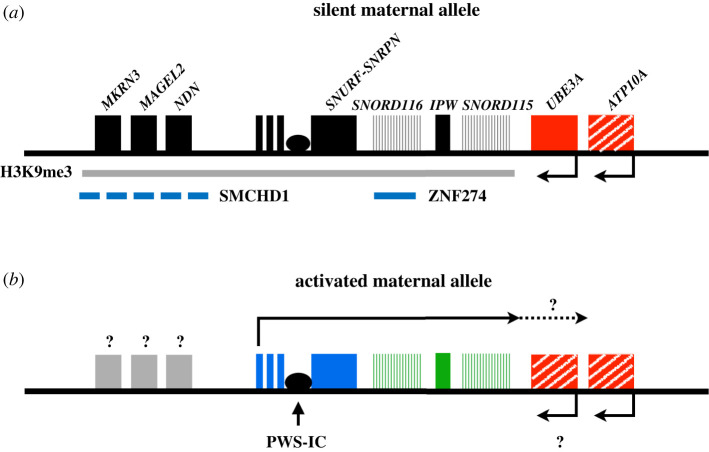
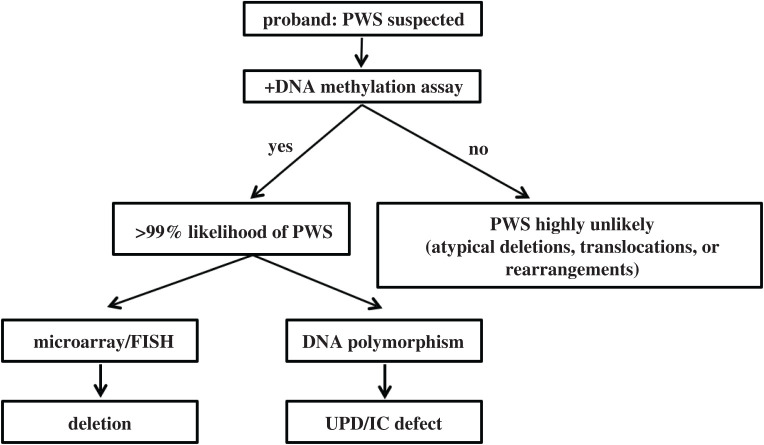
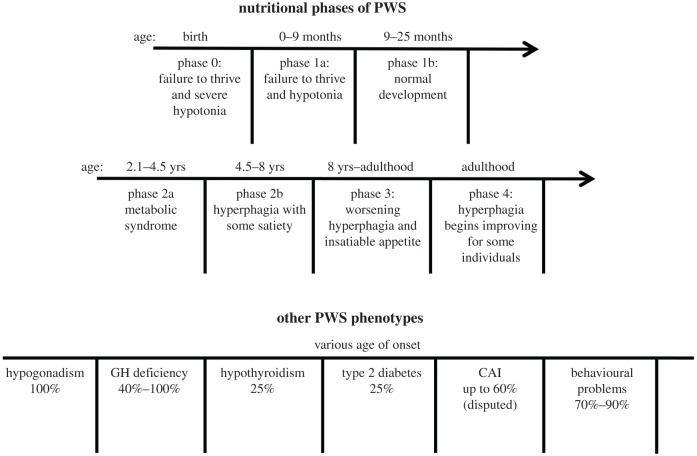
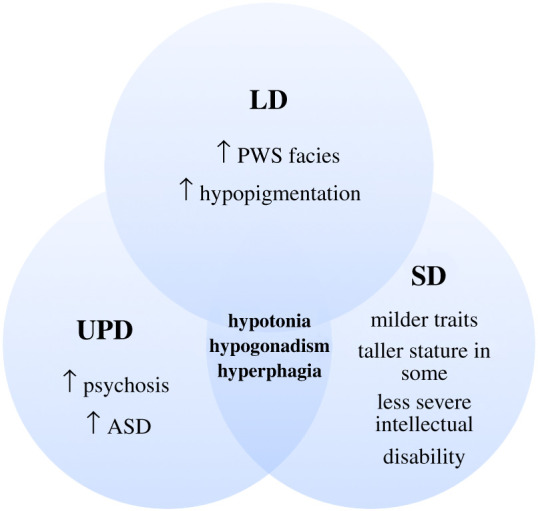
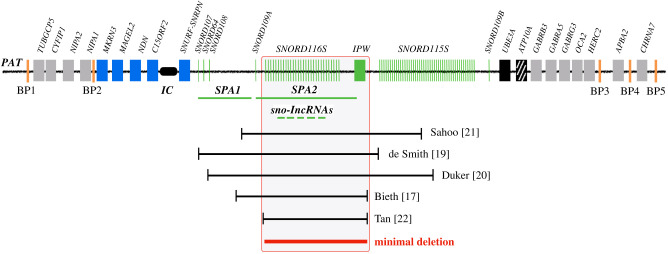
Prader-Willi syndrome (PWS) is caused by the loss of function of the paternally inherited 15q11-q13 locus. This region is governed by genomic imprinting, a phenomenon in which genes are expressed exclusively from one parental allele. The genomic imprinting of the 15q11-q13 locus is established in the germline and is largely controlled by a bipartite imprinting centre. One part, termed the Prader-Willi syndrome imprinting center (PWS-IC), comprises a CpG island that is unmethylated on the paternal allele and methylated on the maternal allele. The second part, termed the Angelman syndrome imprinting centre, is required to silence the PWS_IC in the maternal germline. The loss of the paternal contribution of the imprinted 15q11-q13 locus most frequently occurs owing to a large deletion of the entire imprinted region but can also occur through maternal uniparental disomy or an imprinting defect. While PWS is considered a contiguous gene syndrome based on large-deletion and uniparental disomy patients, the lack of expression of only non-coding RNA transcripts from the may be the primary cause of PWS. Patients with small atypical deletions of the paternal cluster alone appear to have most of the PWS related clinical phenotypes. The loss of the maternal contribution of the 15q11-q13 locus causes a separate and distinct condition called Angelman syndrome. Importantly, while much has been learned about the regulation and expression of genes and transcripts deriving from the 15q11-q13 locus, there remains much to be learned about how these genes and transcripts contribute at the molecular level to the clinical traits and developmental aspects of PWS that have been observed.
普拉德-威利综合征(PWS)是由父系遗传的 15q11-q13 位点功能丧失引起的。该区域受基因组印记调控,即基因仅从一个亲本等位基因表达。15q11-q13 位点的基因组印记在生殖系中建立,并主要受二部分印迹中心控制。一部分称为普拉德-威利综合征印迹中心(PWS-IC),由一个 CpG 岛组成,该岛在父系等位基因上未甲基化,而在母系等位基因上甲基化。第二部分称为 Angelman 综合征印迹中心,需要在母系生殖系中沉默 PWS_IC。印迹的 15q11-q13 位点的父系贡献的缺失最常由于整个印迹区域的大片段缺失而发生,但也可以通过母系单亲二体或印迹缺陷发生。虽然 PWS 被认为是基于大片段缺失和单亲二体的连续基因综合征,但仅从非编码 RNA 转录本中缺乏表达可能是 PWS 的主要原因。单独具有父系 簇小的非典型缺失的患者似乎具有大多数与 PWS 相关的临床表型。15q11-q13 位点母系贡献的缺失导致一种称为 Angelman 综合征的独立且不同的病症。重要的是,尽管已经了解了 15q11-q13 位点基因和转录本的调控和表达,但仍需要更多地了解这些基因和转录本如何在分子水平上为观察到的 PWS 的临床特征和发育方面做出贡献。