Al Rahim Md, Yoon Yonejung, Dimovasili Christina, Shao Zhiping, Huang Qian, Zhang Emily, Kezunovic Nebojsa, Chen Lei, Schaffner Adam, Huntley George W, Ubarretxena-Belandia Iban, Georgakopoulos Anastasios, Robakis Nikolaos K
Departments of Psychiatry and Neuroscience, Center for Molecular Biology and Genetics of Neurodegeneration, Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Nash Family Department of Neuroscience, and the Friedman Brain Institute, The Icahn School of Medicine at Mount Sinai, New York, NY, USA.
Brain Commun. 2020 Jul 20;2(2):fcaa100. doi: 10.1093/braincomms/fcaa100. eCollection 2020.
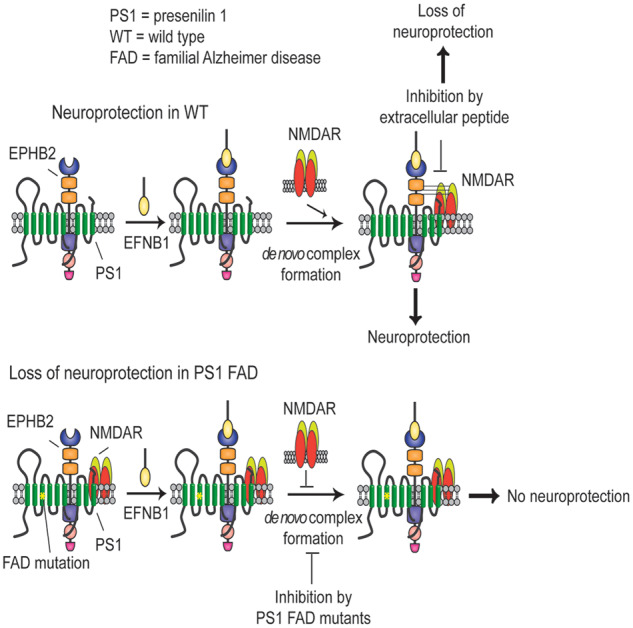
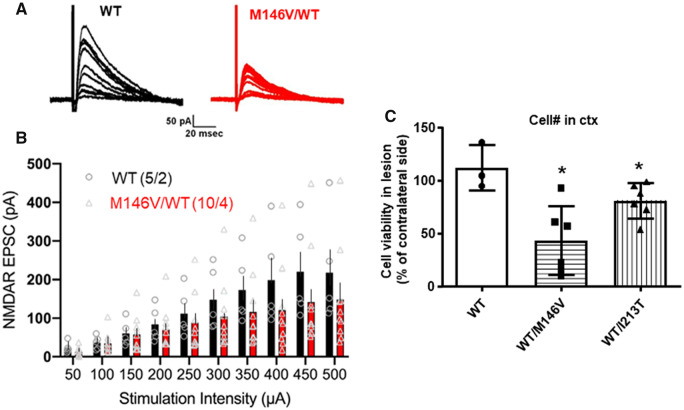
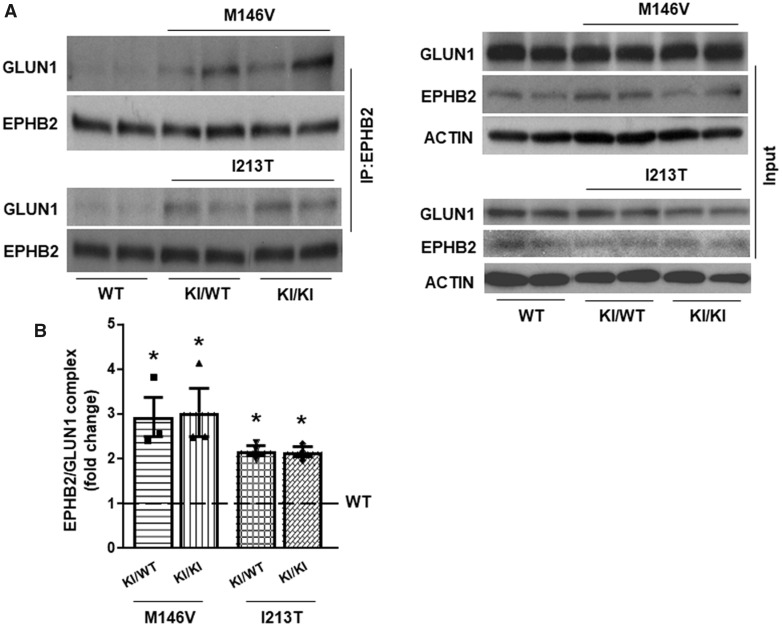
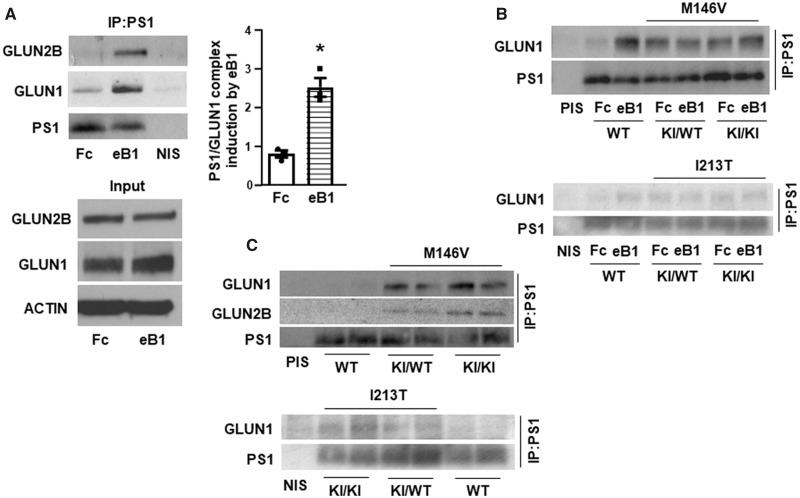
Excitotoxicity is thought to play key roles in brain neurodegeneration and stroke. Here we show that neuroprotection against excitotoxicity by trophic factors EFNB1 and brain-derived neurotrophic factor (called here factors) requires formation of 'survival complexes' which are factor-stimulated complexes of -methyl-d-aspartate receptor with factor receptor and presenilin 1. Absence of presenilin 1 reduces the formation of survival complexes and abolishes neuroprotection. EPH receptor B2- and -methyl-d-aspartate receptor-derived peptides designed to disrupt formation of survival complexes also decrease the factor-stimulated neuroprotection. Strikingly, factor-dependent neuroprotection and levels of the factor-stimulated survival complexes decrease dramatically in neurons expressing presenilin 1 familial Alzheimer disease mutants. Mouse neurons and brains expressing presenilin 1 familial Alzheimer disease mutants contain increased amounts of constitutive presenilin 1--methyl-d-aspartate receptor complexes unresponsive to factors. Interestingly, the stability of the familial Alzheimer disease presenilin 1--methyl-d-aspartate receptor complexes differs from that of wild type complexes and neurons of mutant-expressing brains are more vulnerable to cerebral ischaemia than neurons of wild type brains. Furthermore, -methyl-d-aspartate receptor-mediated excitatory post-synaptic currents at CA1 synapses are altered by presenilin 1 familial Alzheimer disease mutants. Importantly, high levels of presenilin 1--methyl-d-aspartate receptor complexes are also found in post-mortem brains of Alzheimer disease patients expressing presenilin 1 familial Alzheimer disease mutants. Together, our data identify a novel presenilin 1-dependent neuroprotective mechanism against excitotoxicity and indicate a pathway by which presenilin 1 familial Alzheimer disease mutants decrease factor-depended neuroprotection against excitotoxicity and ischaemia in the absence of Alzheimer disease neuropathological hallmarks which may form downstream of neuronal damage. These findings have implications for the pathogenic effects of familial Alzheimer disease mutants and therapeutic strategies.
兴奋性毒性被认为在脑神经元变性和中风中起关键作用。在此我们表明,神经营养因子EFNB1和脑源性神经营养因子(在此称为因子)对兴奋性毒性的神经保护作用需要形成“存活复合物”,即因子刺激的N-甲基-D-天冬氨酸受体与因子受体和早老素1的复合物。早老素1的缺失会减少存活复合物的形成并消除神经保护作用。设计用于破坏存活复合物形成的EPH受体B2和N-甲基-D-天冬氨酸受体衍生肽也会降低因子刺激的神经保护作用。令人惊讶的是,在表达早老素1家族性阿尔茨海默病突变体的神经元中,因子依赖性神经保护作用和因子刺激的存活复合物水平显著降低。表达早老素1家族性阿尔茨海默病突变体的小鼠神经元和大脑中,组成性早老素1-N-甲基-D-天冬氨酸受体复合物的含量增加,且对因子无反应。有趣的是,家族性阿尔茨海默病早老素1-N-甲基-D-天冬氨酸受体复合物的稳定性与野生型复合物不同,表达突变体的大脑中的神经元比野生型大脑中的神经元更容易受到脑缺血的影响。此外,早老素1家族性阿尔茨海默病突变体改变了CA1突触处N-甲基-D-天冬氨酸受体介导的兴奋性突触后电流。重要的是,在表达早老素1家族性阿尔茨海默病突变体的阿尔茨海默病患者的死后大脑中也发现了高水平的早老素1-N-甲基-D-天冬氨酸受体复合物。总之,我们的数据确定了一种新的早老素1依赖性抗兴奋性毒性的神经保护机制,并指出了一条途径,通过该途径,早老素1家族性阿尔茨海默病突变体在没有可能在神经元损伤下游形成的阿尔茨海默病神经病理特征的情况下,降低了因子依赖性抗兴奋性毒性和缺血的神经保护作用。这些发现对家族性阿尔茨海默病突变体的致病作用和治疗策略具有重要意义。