Institute for Medical Microbiology, Virology and Hygiene, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany.
Heinrich Pette Institute, Leibniz Institute for Experimental Virology, Virus Genomics, Hamburg, Germany.
Clin Microbiol Infect. 2021 Jan;27(1):130.e5-130.e8. doi: 10.1016/j.cmi.2020.09.034. Epub 2020 Sep 29.
Investigation whether in depth characterization of virus variant patterns can be used for epidemiological analysis of the first severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection clusters in Hamburg, Germany.
Metagenomic RNA-sequencing and amplicon-sequencing and subsequent variant calling in 25 respiratory samples from SARS-CoV-2 infected patients involved in the earliest infection clusters in Hamburg.
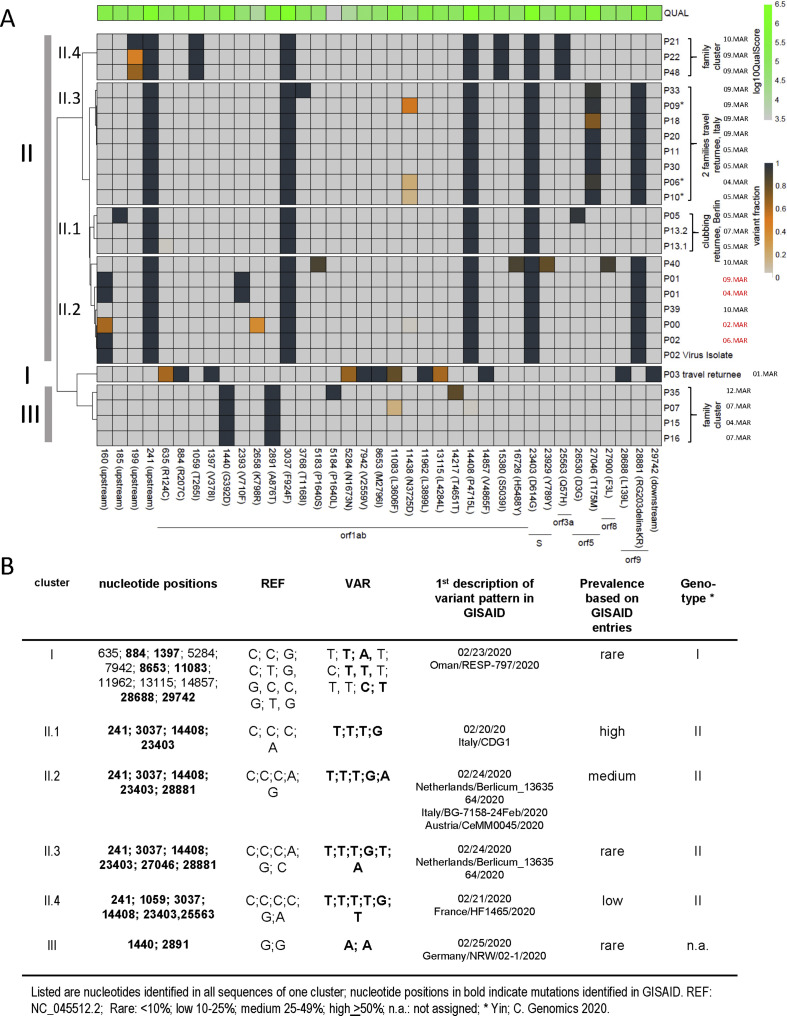
Amplikon sequencing and cluster analyses of these SARS-CoV-2 sequences allowed the identification of the first infection cluster and five non-related infection clusters occurring at the beginning of the viral entry of SARS-CoV-2 in the Hamburg metropolitan region. Viral genomics together with epidemiological analyses revealed that the index patient acquired the infection in northern Italy and transmitted it to two out of 134 contacts. Single nucleotide polymorphisms clearly distinguished the virus variants of the index and other clusters and allowed us to track in which sequences worldwide these mutations were first described. Minor variant analyses identified the transmission of intra-host variants in the index cluster and household clusters.
SARS-CoV-2 variant tracing allows the identification of infection clusters and the follow up of infection chains occurring in the population. Furthermore, the follow up of minor viral variants in infection clusters can provide further resolution on transmission events indistinguishable at a consensus sequence level.
探究深入分析病毒变异模式是否可用于对德国汉堡首次严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)感染群进行流行病学分析。
对来自参与汉堡最早感染群的 25 名 SARS-CoV-2 感染患者的 25 份呼吸道样本进行宏基因组 RNA 测序和扩增子测序,并随后进行变异调用。
对这些 SARS-CoV-2 序列进行扩增子测序和聚类分析,可识别出首个感染群和在 SARS-CoV-2 进入汉堡大都市区初期发生的五个不相关的感染群。病毒基因组学分析结合流行病学分析表明,该指数患者在意大利北部感染了病毒,并将其传播给了 134 名接触者中的 2 人。单核苷酸多态性清楚地区分了指数和其他集群的病毒变体,并使我们能够追踪这些突变首次在世界范围内哪些序列中被描述。微小变体分析确定了在指数集群和家庭集群中宿主内变体的传播。
SARS-CoV-2 变体追踪可识别感染群,并跟踪人群中发生的感染链。此外,对感染群中微小病毒变体的跟踪可以在共识序列水平上无法区分的传播事件提供进一步的分辨率。