National Reference Centre (NRC) for Whole Genome Sequencing of microbial pathogens: data-base and bioinformatics analysis (GENPAT), Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise "Giuseppe Caporale" (IZSAM), via Campo Boario, 64100, Teramo, TE, Italy.
BMC Genomics. 2021 Oct 30;22(1):782. doi: 10.1186/s12864-021-08112-0.
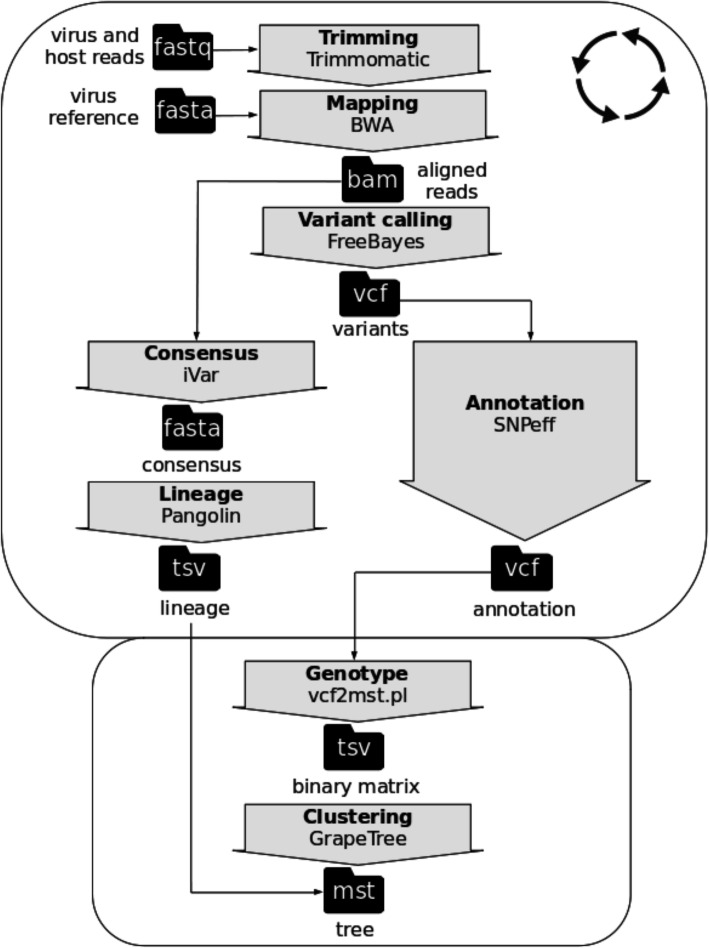
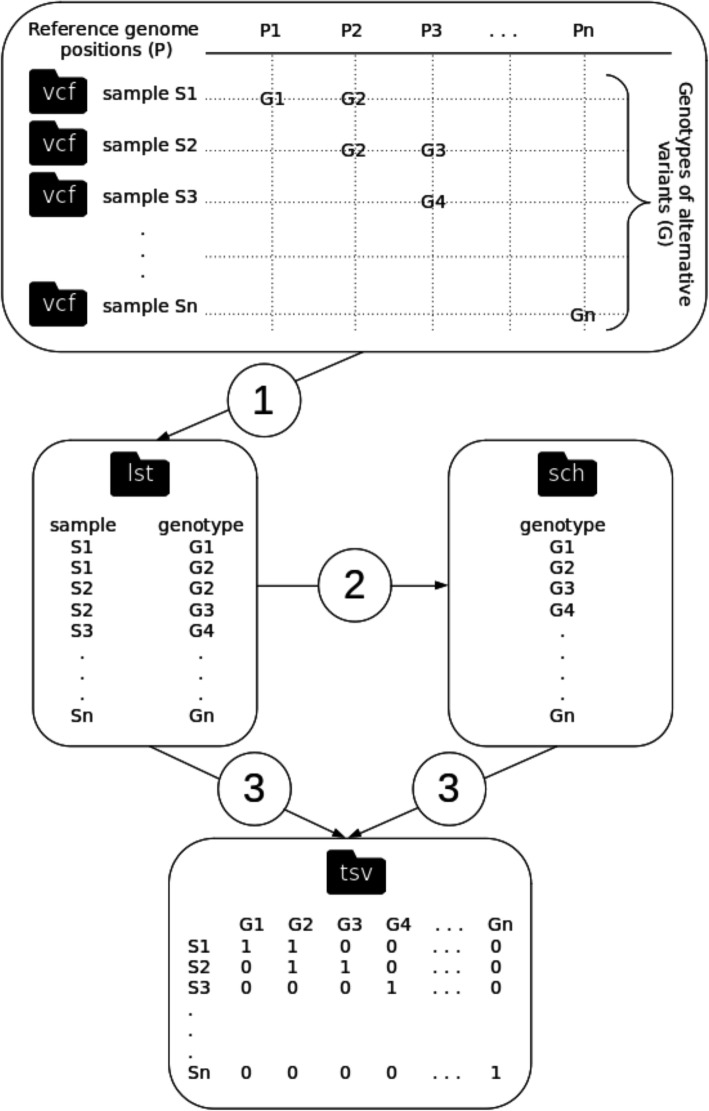
Faced with the ongoing global pandemic of coronavirus disease, the 'National Reference Centre for Whole Genome Sequencing of microbial pathogens: database and bioinformatic analysis' (GENPAT) formally established at the 'Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise' (IZSAM) in Teramo (Italy) is in charge of the SARS-CoV-2 surveillance at the genomic scale. In a context of SARS-CoV-2 surveillance requiring correct and fast assessment of epidemiological clusters from substantial amount of samples, the present study proposes an analytical workflow for identifying accurately the PANGO lineages of SARS-CoV-2 samples and building of discriminant minimum spanning trees (MST) bypassing the usual time consuming phylogenomic inferences based on multiple sequence alignment (MSA) and substitution model.
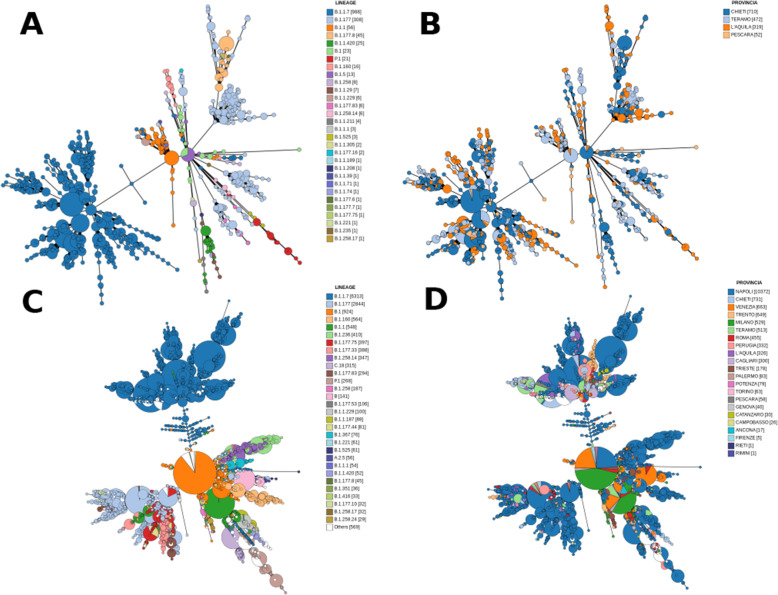
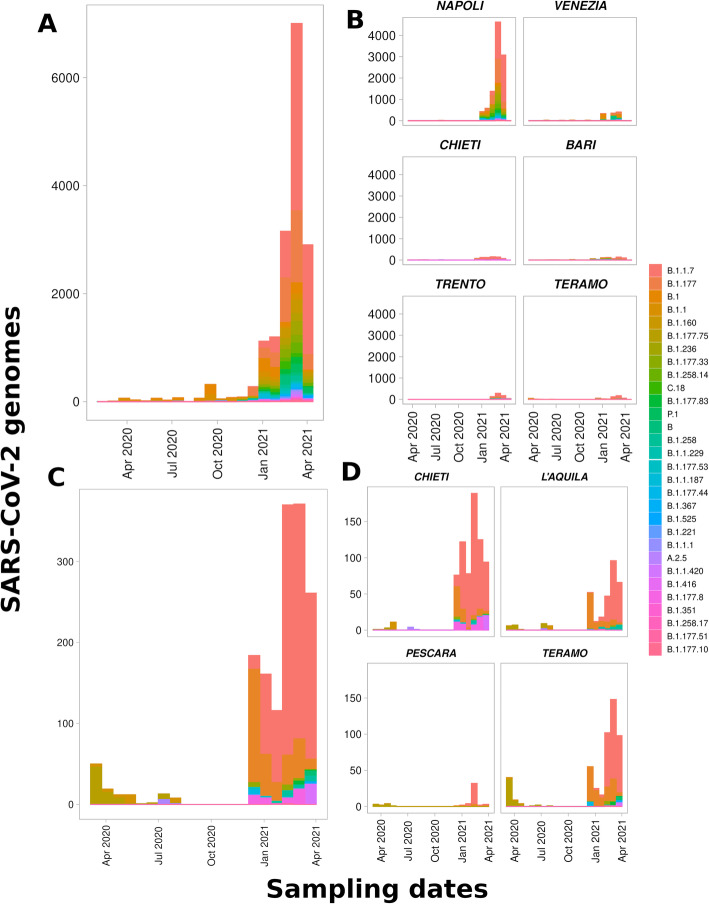
GENPAT constituted two collections of SARS-CoV-2 samples. The first collection consisted of SARS-CoV-2 positive swabs collected by IZSAM from the Abruzzo region (Italy), then sequenced by next generation sequencing (NGS) and analyzed in GENPAT (n = 1592), while the second collection included samples from several Italian provinces and retrieved from the reference Global Initiative on Sharing All Influenza Data (GISAID) (n = 17,201). The main results of the present work showed that (i) GENPAT and GISAID detected the same PANGO lineages, (ii) the PANGO lineages B.1.177 (i.e. historical in Italy) and B.1.1.7 (i.e. 'UK variant') are major concerns today in several Italian provinces, and the new MST-based method (iii) clusters most of the PANGO lineages together, (iv) with a higher dicriminatory power than PANGO lineages, (v) and faster that the usual phylogenomic methods based on MSA and substitution model.
The genome sequencing efforts of Italian provinces, combined with a structured national system of NGS data management, provided support for surveillance SARS-CoV-2 in Italy. We propose to build phylogenomic trees of SARS-CoV-2 variants through an accurate, discriminant and fast MST-based method avoiding the typical time consuming steps related to MSA and substitution model-based phylogenomic inference.
面对冠状病毒病这一持续的全球大流行,位于意大利特兰托的“全基因组测序微生物病原体国家参考中心:数据库和生物信息分析”(GENPAT)正式成立,负责在基因组范围内进行 SARS-CoV-2 监测。在 SARS-CoV-2 监测的背景下,需要对大量样本的流行病学集群进行正确快速的评估,本研究提出了一种分析工作流程,用于准确识别 SARS-CoV-2 样本的 PANGO 谱系,并构建判别最小生成树(MST),绕过基于多序列比对(MSA)和替代模型的耗时的系统发育推断。
GENPAT 由两部分 SARS-CoV-2 样本组成。第一部分是由 IZSAM 从意大利阿布鲁佐地区采集的 SARS-CoV-2 阳性拭子组成,然后通过下一代测序(NGS)进行测序,并在 GENPAT 中进行分析(n=1592),而第二部分包括来自意大利几个省份的样本,并从参考全球流感共享倡议数据(GISAID)中检索(n=17201)。本工作的主要结果表明:(i)GENPAT 和 GISAID 检测到相同的 PANGO 谱系;(ii)谱系 B.1.177(即在意大利的历史谱系)和 B.1.1.7(即“英国变体”)是目前意大利几个省份的主要关注点;(iii)新的基于 MST 的方法将大多数 PANGO 谱系聚类在一起;(iv)比 PANGO 谱系具有更高的辨别力;(v)比基于 MSA 和替代模型的典型系统发育推断方法更快。
意大利各省的基因组测序工作,加上结构化的国家 NGS 数据管理系统,为意大利的 SARS-CoV-2 监测提供了支持。我们建议通过基于准确、有辨别力和快速 MST 的方法构建 SARS-CoV-2 变体的系统发育树,避免与基于 MSA 和替代模型的系统发育推断相关的典型耗时步骤。