Jing Ren, Hu Zhao-Kun, Lin Fei, He Sheng, Zhang Sui-Sui, Ge Wan-Yun, Dai Hui-Jun, Du Xue-Ke, Lin Jin-Yuan, Pan Ling-Hui
Department of Anesthesiology, Guangxi Medical University Affiliated Tumor Hospital & Oncology Medical College, Nanning, China.
The Laboratory of Perioperative Medicine Research Center, Guangxi Medical University Affiliated Tumor Hospital & Oncology Medical College, Nanning, China.
Front Cell Dev Biol. 2020 Sep 4;8:819. doi: 10.3389/fcell.2020.00819. eCollection 2020.
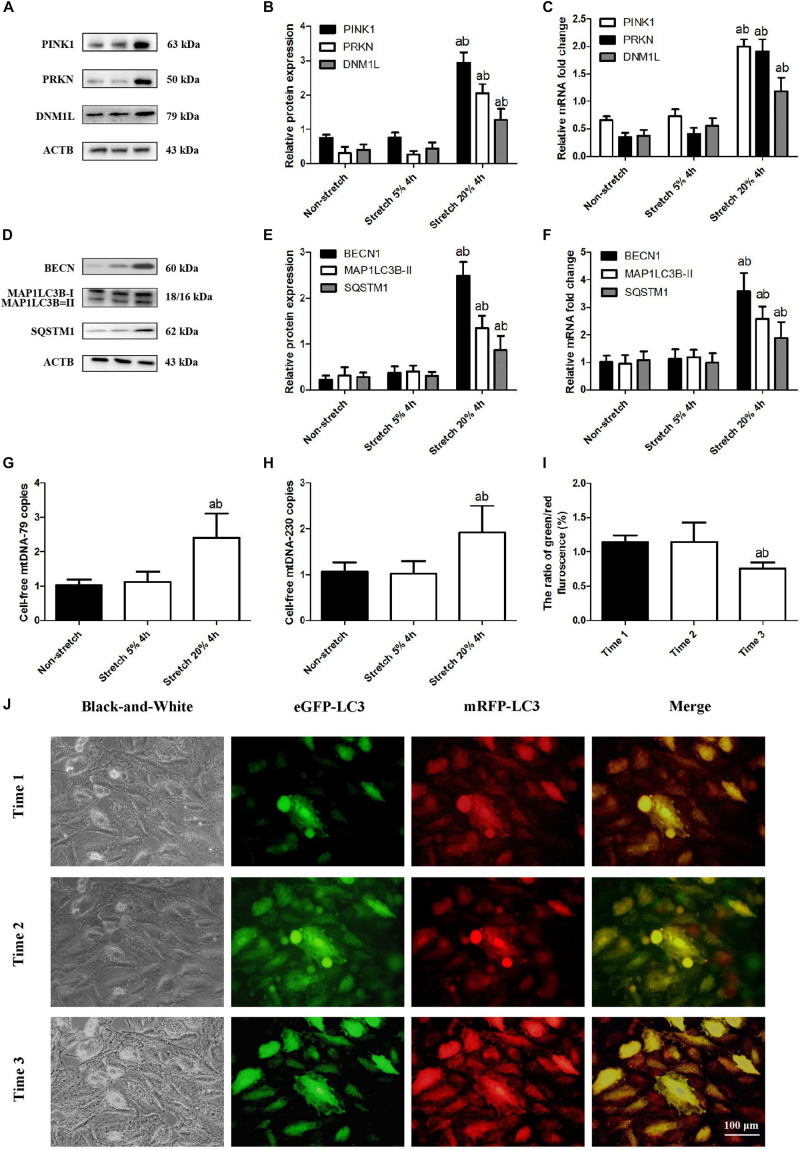
In animal models of ventilation-induced lung injury, mitophagy triggers mitochondria damage and the release of mitochondrial (mt) DNA, which activates inflammation. However, the mechanism of this process is unclear.
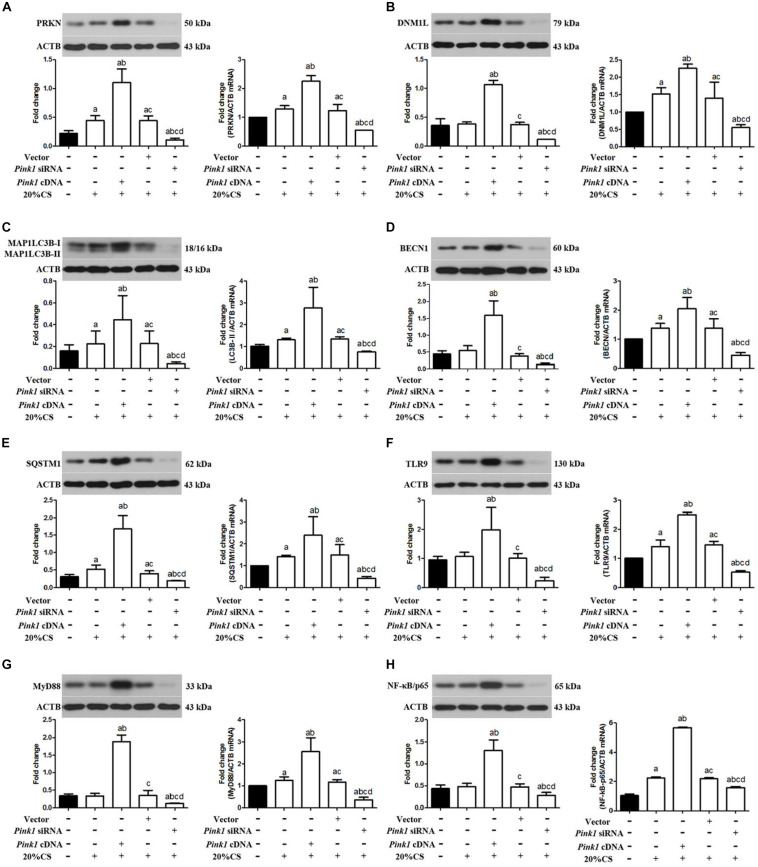
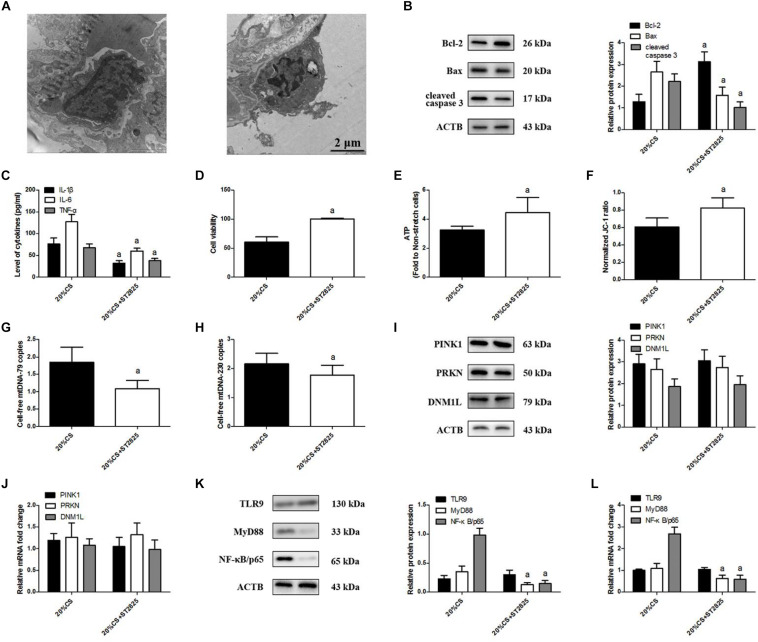
A model of cyclic stretching (CS)-induced lung epithelial cell injury was established. The genetic intervention of phosphatase and tensin homolog-induced kinase 1 (PINK1) expression via lentivirus transfection was used to identify the relationship between PINK1-mediated mitophagy and mtDNA release in stretching-induced inflammatory response and injury. Pharmacological inhabitation of Toll-like receptor 9 (TLR9) and myeloid differentiation factor 88 (MyD88) expression was performed via their related inhibitors, while pre-treatment of exogenous mtDNA was used to verify the role of mtDNA in stretching-induced inflammatory response and injury.
Using a cell culture model of CS, we found that knocking down PINK1 in lung epithelial cells reduced mitophagy activation and mtDNA release, leading to milder inflammatory response and injury; conversely, up-regulating PINK1 exacerbated stretching-induced inflammation and injury, and similar effects were observed by upregulating TLR9 to induce expression of MyD88 and nuclear factor-κB (NF-κB)/p65. Down-regulating MyD88 protected lung epithelial cells from stretching injury and decreased NF-κB/p65 expression.
These findings suggest that PINK1-dependent mitophagy and associated TLR9 activation is indeed a major factor in stretch-induced cell injury via a mechanism in which released mtDNA activates TLR9 and thereby the MyD88/NF-κB pathway. Inhibiting this process may be a therapeutic approach to prevent inflammation and cell injury in patients on mechanical ventilation.
在机械通气诱导的肺损伤动物模型中,线粒体自噬引发线粒体损伤并释放线粒体(mt)DNA,进而激活炎症反应。然而,这一过程的机制尚不清楚。
建立循环拉伸(CS)诱导的肺上皮细胞损伤模型。通过慢病毒转染对磷酸酶和张力蛋白同源物诱导激酶1(PINK1)表达进行基因干预,以确定PINK1介导的线粒体自噬与拉伸诱导的炎症反应和损伤中mtDNA释放之间的关系。通过相关抑制剂对Toll样受体9(TLR9)和髓样分化因子88(MyD88)表达进行药理学抑制,同时用外源性mtDNA预处理以验证mtDNA在拉伸诱导的炎症反应和损伤中的作用。
利用CS细胞培养模型,我们发现敲低肺上皮细胞中的PINK1可减少线粒体自噬激活和mtDNA释放,导致炎症反应和损伤减轻;相反,上调PINK1会加剧拉伸诱导的炎症和损伤,上调TLR9以诱导MyD88和核因子-κB(NF-κB)/p65表达也观察到类似效果。下调MyD88可保护肺上皮细胞免受拉伸损伤并降低NF-κB/p65表达。
这些发现表明,PINK1依赖的线粒体自噬和相关的TLR9激活确实是拉伸诱导细胞损伤的主要因素,其机制是释放的mtDNA激活TLR9,从而激活MyD88/NF-κB途径。抑制这一过程可能是预防机械通气患者炎症和细胞损伤的一种治疗方法。