Khani Farnousha, Atabaki Hooshang
Department of Chemistry, Arak Branch, Islamic Azad University, Arak, Iran.
ACS Omega. 2020 Sep 18;5(38):24311-24317. doi: 10.1021/acsomega.0c02506. eCollection 2020 Sep 29.
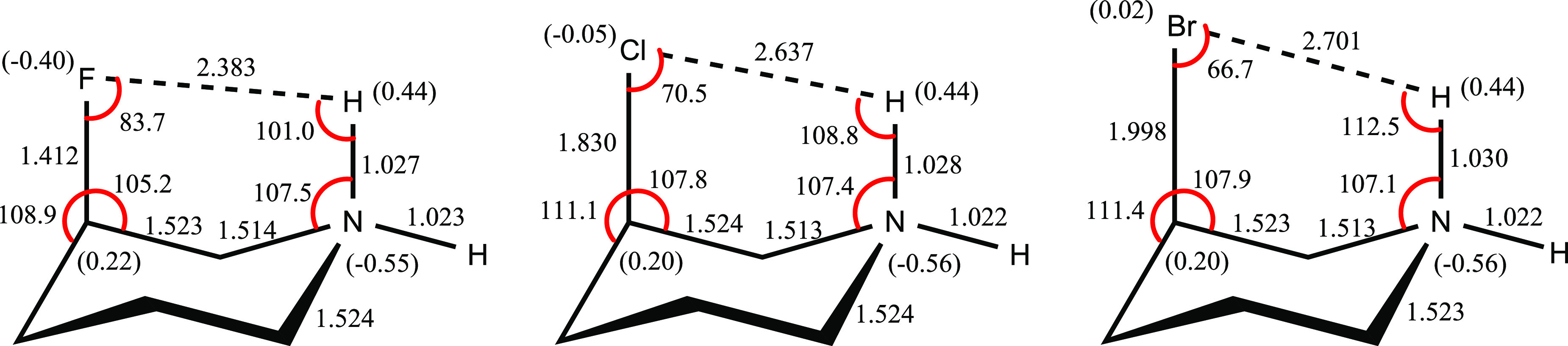
Although there are some published conclusions in the literature concerning the origin of the axial-conformation preference in 3-fluoropiperidinium cations (charge-dipole orientation effect), the origin of the axial-conformation preferences in the 3-halopiperidinium cations [halogen = F (), Cl (), Br ()] has remained an open question. To explore the origin of the axial-conformation preferences in compounds -, we assessed the roles and contributions of the hyperconjugative interactions, the Coulombic electrostatic interactions, the electrostatic model associated with dipole-dipole interactions, and the steric effects associated with the Pauli exchange-type repulsions on the conformational properties of compounds - utilizing the G3MP2, LC-ωPBE, and B3LYP methods and natural bond orbital (NBO) interpretations. Natural Coulombic potential energies are in favor of the axial conformations of compounds -, and justify their corresponding total energy differences. The through-space hyperconjugative interactions between the donor lone pairs of halogen atoms (LPX) and the acceptor antibonding orbitals of H-N bonds [σ* ], LPX → σ* , increase from compound to compound . The inspection of the dipole moments of the parallel C-X and H-N bonds in the axial conformations of compounds - revealed that the variations of their corresponding four-center dipole-dipole interactions correlate well with their corresponding conformational behaviors. The steric effects associated with the Pauli exchange-type repulsions are strongly in favor of the equatorial conformations of compounds -. Accordingly, the charge-dipole orienting effect associated with the four-center dipole-dipole interactions is a dominant factor in the conformational behaviors of compounds -.
尽管文献中已有一些关于3-氟哌啶鎓阳离子轴向构象偏好起源(电荷-偶极取向效应)的结论,但3-卤代哌啶鎓阳离子[卤素 = F()、Cl()、Br()]轴向构象偏好的起源仍是一个悬而未决的问题。为了探究化合物 - 的轴向构象偏好起源,我们利用G3MP2、LC-ωPBE和B3LYP方法以及自然键轨道(NBO)解释,评估了超共轭相互作用、库仑静电相互作用、与偶极-偶极相互作用相关的静电模型以及与泡利交换型排斥相关的空间效应在化合物 - 构象性质上的作用和贡献。自然库仑势能有利于化合物 - 的轴向构象,并证明了它们相应的总能量差异。卤素原子的供体孤对电子(LPX)与H-N键的受体反键轨道[σ*]之间的空间超共轭相互作用,LPX→σ*,从化合物 到化合物 逐渐增加。对化合物 - 轴向构象中平行的C-X和H-N键的偶极矩进行检查发现,它们相应的四中心偶极-偶极相互作用的变化与其相应的构象行为密切相关。与泡利交换型排斥相关的空间效应强烈有利于化合物 - 的赤道构象。因此,与四中心偶极-偶极相互作用相关的电荷-偶极取向效应是化合物 - 构象行为的主导因素。