Ke Honggang, Wu Yunyu, Wang Runjie, Wu Xiaohong
Department of Cardiovascular and Thoracic Surgery, Affiliated Hospital of Nantong University, Nantong, Jiangsu, China (mainland).
Qixiu Campus, Nantong University, Nantong, Jiangsu, China (mainland).
Med Sci Monit. 2020 Oct 6;26:e925833. doi: 10.12659/MSM.925833.
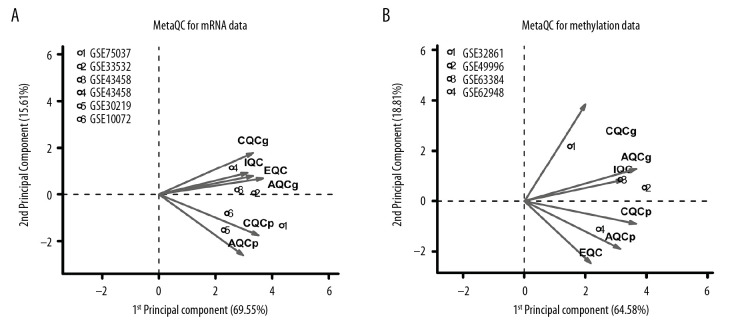
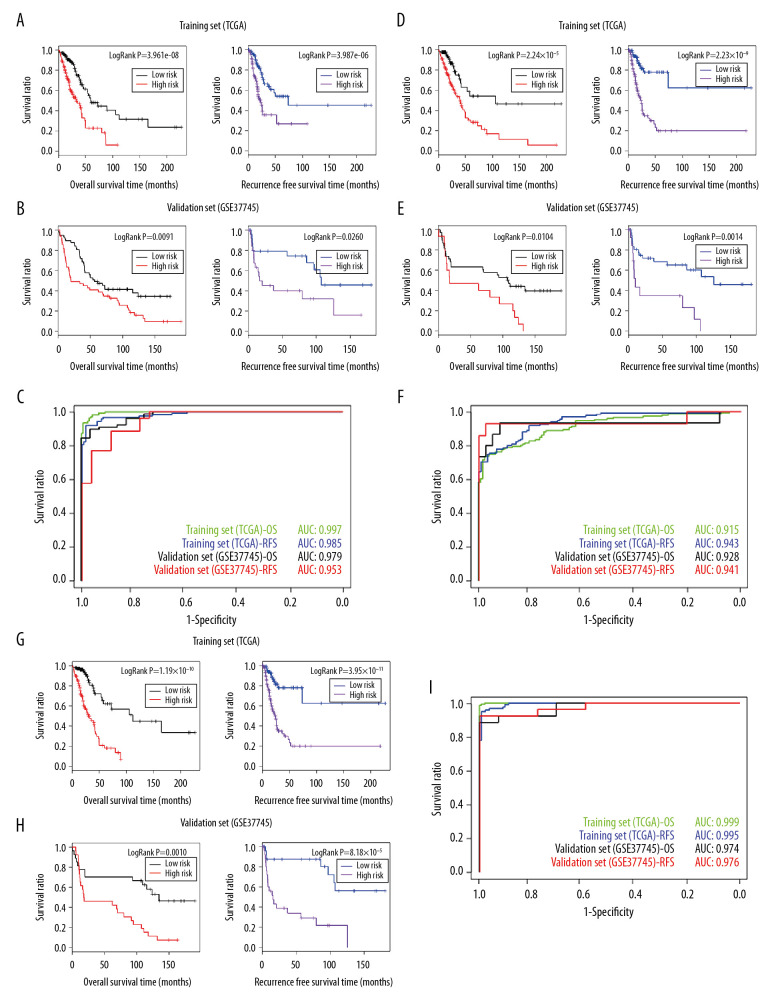
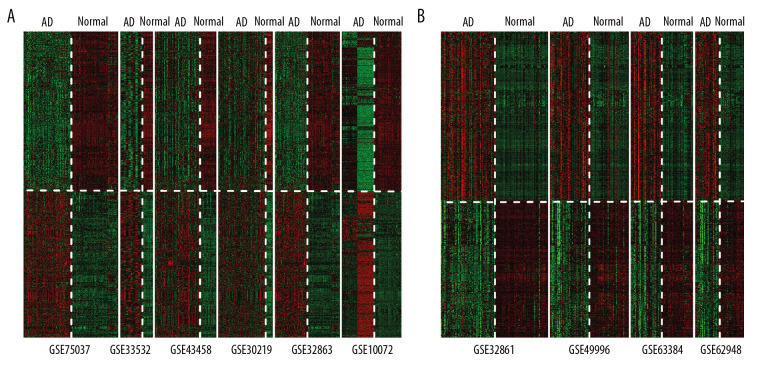
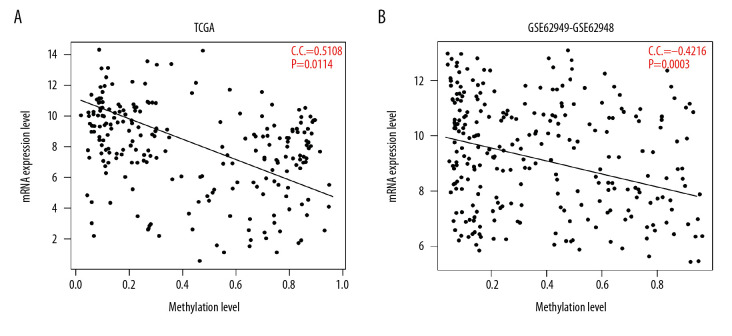
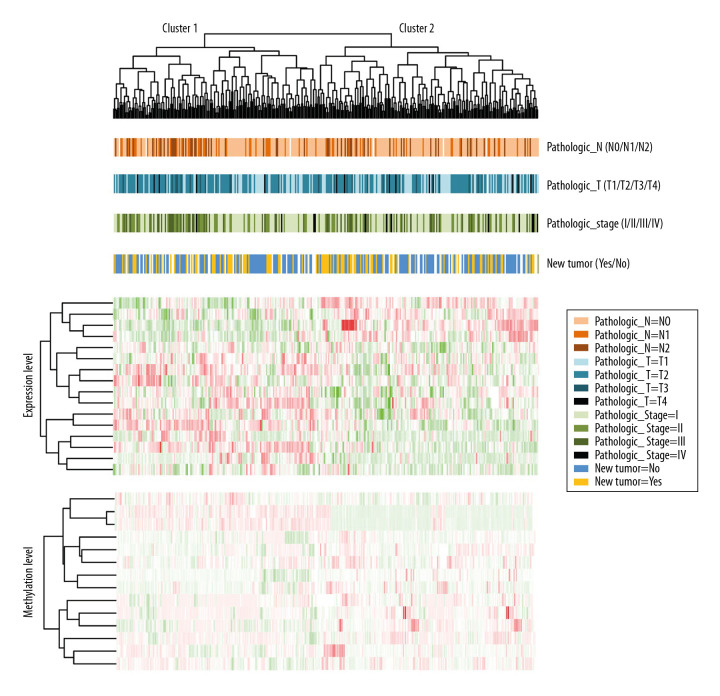
BACKGROUND This study aimed to identify important marker genes in lung adenocarcinoma (LACC) and establish a prognostic risk model to predict the risk of LACC in patients. MATERIAL AND METHODS Gene expression and methylation profiles for LACC and clinical information about cases were downloaded from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases, respectively. Differentially expressed genes (DEGs) and differentially methylated genes (DMGs) between cancer and control groups were selected through meta-analysis. Pearson coefficient correlation analysis was performed to identify intersections between DEGs and DMGs and a functional analysis was performed on the genes that were correlated. Marker genes and clinical factors significantly related to prognosis were identified using univariate and multivariate Cox regression analyses. Risk prediction models were then created based on the marker genes and clinical factors. RESULTS In total, 1975 DEGs and 2095 DMGs were identified. After comparison, 16 prognosis-related genes (EFNB2, TSPAN7, INPP5A, VAMP2, CALML5, SNAI2, RHOBTB1, CKB, ATF7IP2, RIMS2, RCBTB2, YBX1, RAB27B, NFATC1, TCEAL4, and SLC16A3) were selected from 265 overlapping genes. Four clinical factors (pathologic N [node], pathologic T [tumor], pathologic stage, and new tumor) were associated with prognosis. The prognostic risk prediction models were constructed and validated with other independent datasets. CONCLUSIONS An integrated model that combines clinical factors and gene markers is useful for predicting risk of LACC in patients. The 16 genes that were identified, including EFNB2, TSPAN7, INPP5A, VAMP2, and CALML5, may serve as novel biomarkers for diagnosis of LACC and prediction of disease prognosis.
背景 本研究旨在鉴定肺腺癌(LACC)中的重要标志物基因,并建立一个预后风险模型以预测患者患LACC的风险。
材料与方法 分别从基因表达综合数据库(GEO)和癌症基因组图谱(TCGA)数据库下载LACC的基因表达和甲基化谱以及病例的临床信息。通过荟萃分析选择癌症组与对照组之间的差异表达基因(DEG)和差异甲基化基因(DMG)。进行Pearson系数相关性分析以鉴定DEG和DMG之间的交集,并对相关基因进行功能分析。使用单因素和多因素Cox回归分析鉴定与预后显著相关的标志物基因和临床因素。然后基于标志物基因和临床因素创建风险预测模型。
结果 共鉴定出1975个DEG和2095个DMG。比较后,从265个重叠基因中选择了16个与预后相关的基因(EFNB2、TSPAN7、INPP5A、VAMP2、CALML5、SNAI2、RHOBTB1、CKB、ATF7IP2、RIMS2、RCBTB2、YBX1、RAB27B、NFATC1、TCEAL4和SLC16A3)。四个临床因素(病理N[淋巴结]、病理T[肿瘤]、病理分期和新肿瘤)与预后相关。构建了预后风险预测模型并用其他独立数据集进行了验证。
结论 结合临床因素和基因标志物的综合模型有助于预测患者患LACC的风险。所鉴定的16个基因,包括EFNB2、TSPAN7、INPP5A、VAMP2和CALML5,可能作为LACC诊断和疾病预后预测的新型生物标志物。