Department of Cellular Biology and Anatomy, Louisiana State University Health Sciences Center, Shreveport, LA, USA.
Department of Toxicology and Cancer Biology, College of Medicine, University of Kentucky, Lexington, KY, USA.
Redox Biol. 2020 Oct;37:101740. doi: 10.1016/j.redox.2020.101740. Epub 2020 Sep 30.
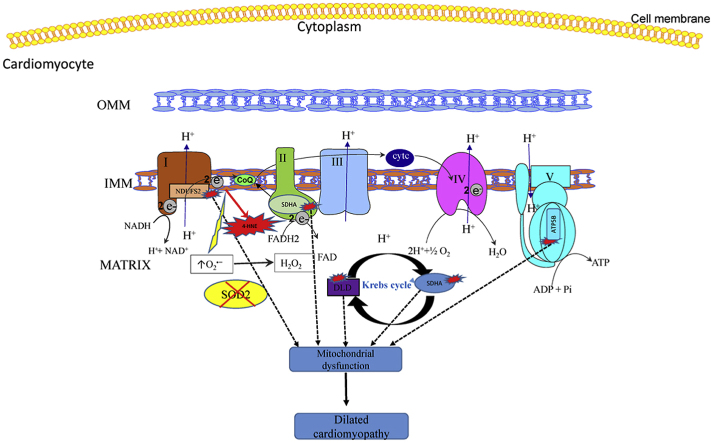
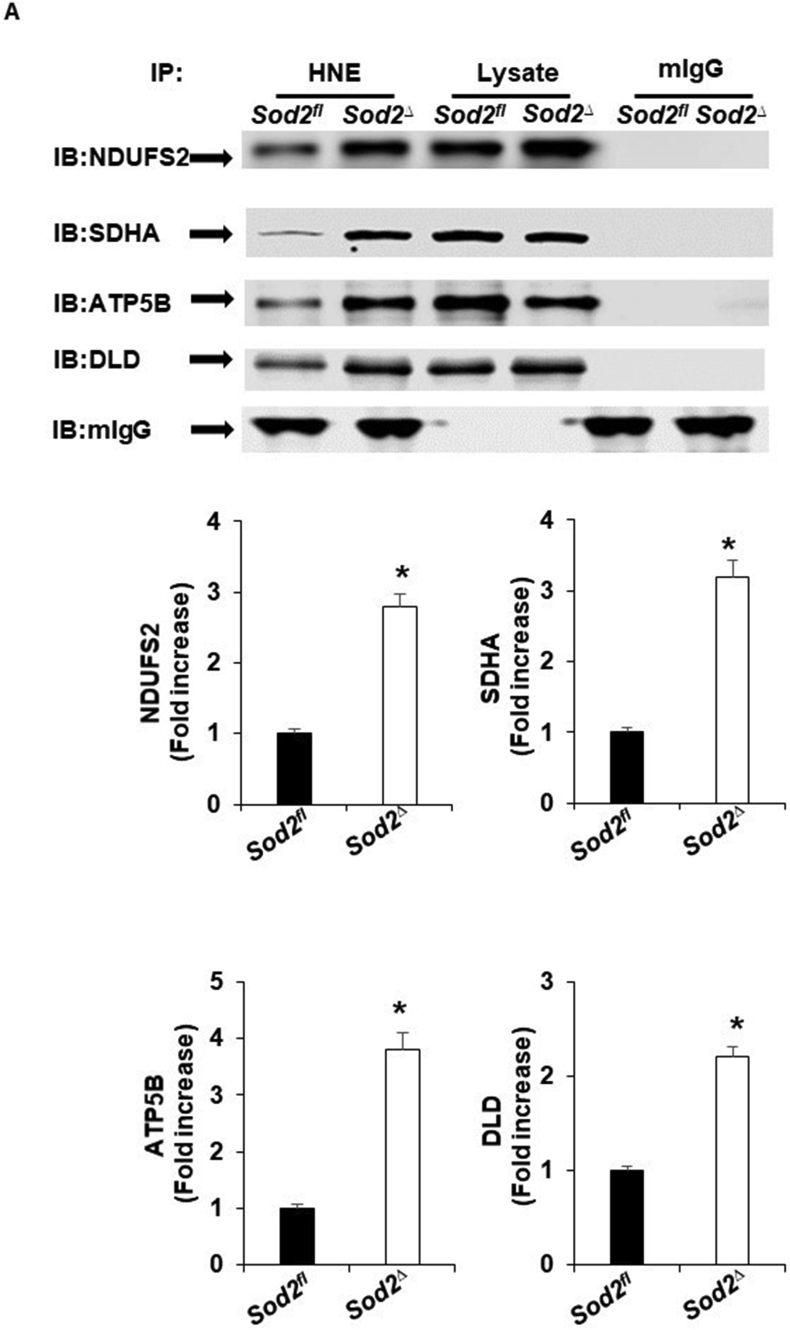
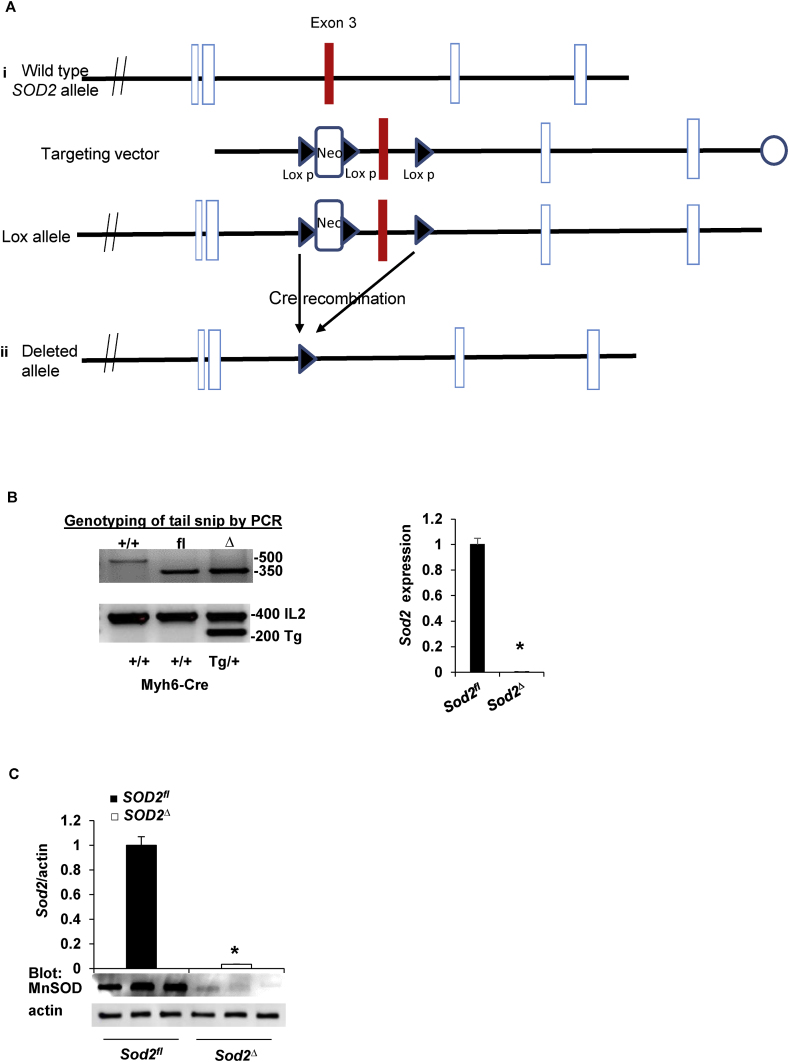
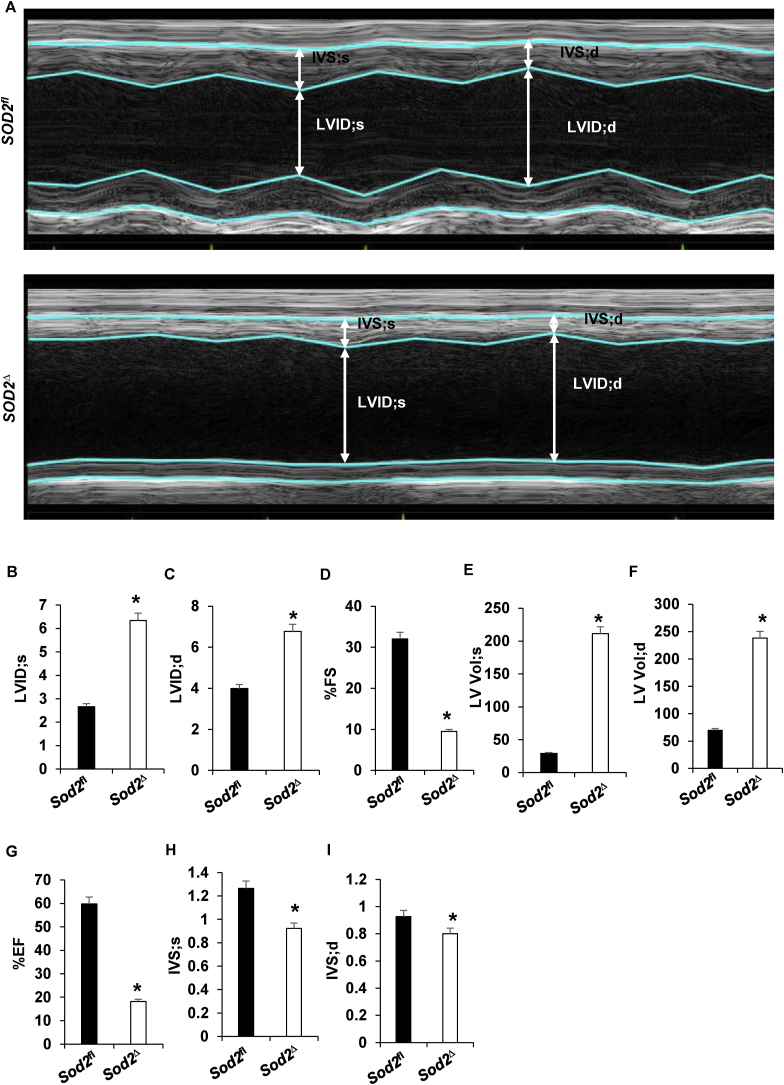
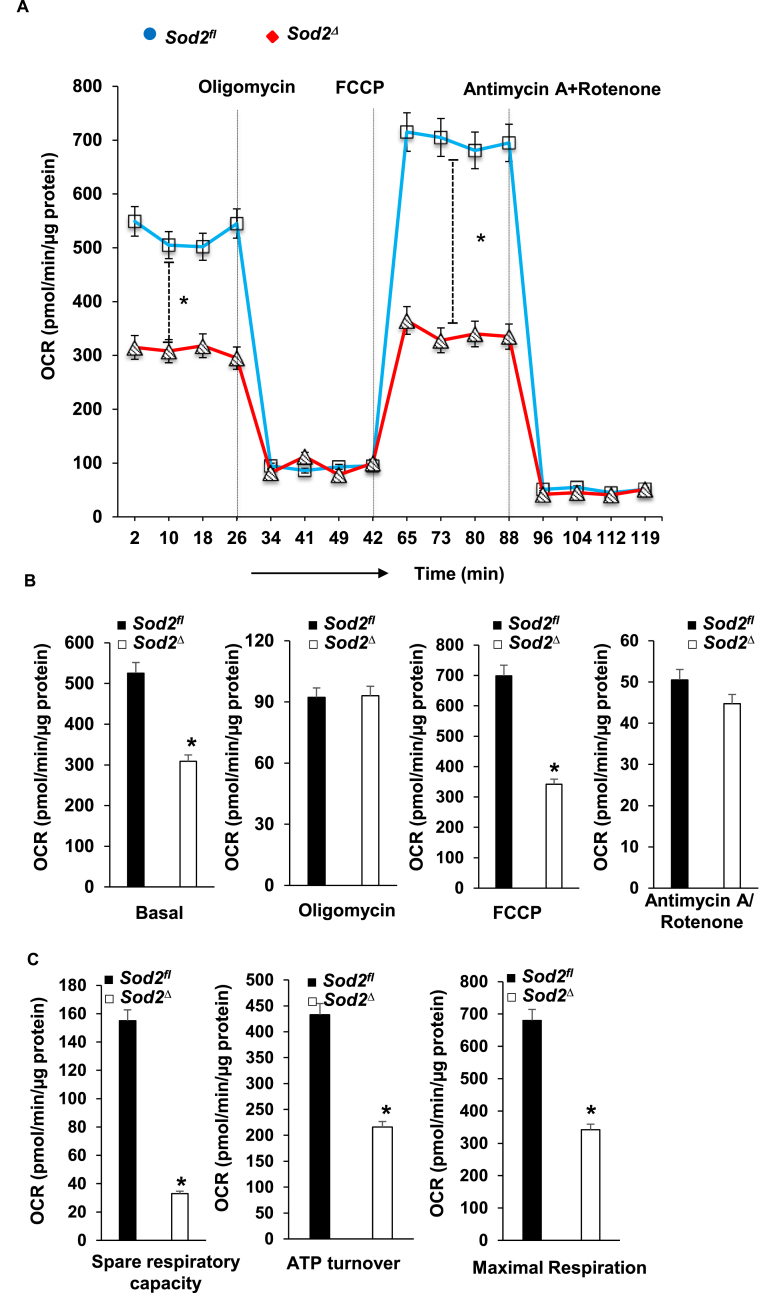
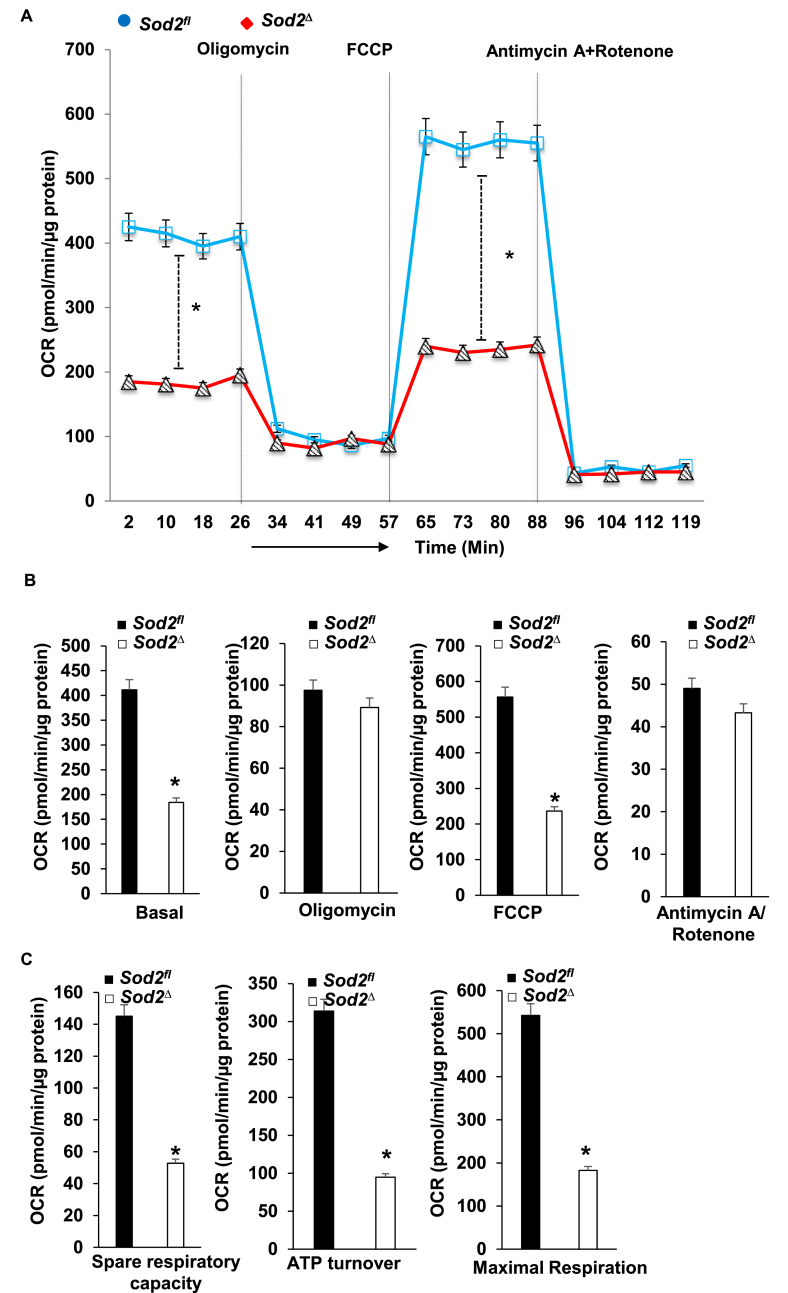
Electrophilic aldehyde (4-hydroxynonenal; 4-HNE), formed after lipid peroxidation, is a mediator of mitochondrial dysfunction and implicated in both the pathogenesis and the progression of cardiovascular disease. Manganese superoxide dismutase (MnSOD), a nuclear-encoded antioxidant enzyme, catalyzes the dismutation of superoxide radicals (O) in mitochondria. To study the role of MnSOD in the myocardium, we generated a cardiomyocyte-specific SOD2 (SOD2Δ) deficient mouse strain. Unlike global SOD2 knockout mice, SOD2Δ mice reached adolescence; however, they die at ~4 months of age due to heart failure. Ultrastructural analysis of SOD2Δ hearts revealed altered mitochondrial architecture, with prominent disruption of the cristae and vacuole formation. Noninvasive echocardiographic measurements in SOD2 mice showed dilated cardiomyopathic features such as decreased ejection fraction and fractional shortening along with increased left ventricular internal diameter. An increased incidence of ventricular tachycardia was observed during electrophysiological studies of the heart in SOD2Δ mice. Oxidative phosphorylation (OXPHOS) measurement using a Seahorse XF analyzer in SOD2Δ neonatal cardiomyocytes and adult cardiac mitochondria displayed reduced O consumption, particularly during basal conditions and after the addition of FCCP (H ionophore/uncoupler), compared to that in SOD2fl hearts. Measurement of extracellular acidification (ECAR) to examine glycolysis in these cells showed a pattern precisely opposite that of the oxygen consumption rate (OCR) among SOD2Δ mice compared to their SOD2 littermates. Analysis of the activity of the electron transport chain complex identified a reduction in Complex I and Complex V activity in SOD2Δ compared to SOD2fl mice. We demonstrated that a deficiency of SOD2 increases reactive oxygen species (ROS), leading to subsequent overproduction of 4-HNE inside mitochondria. Mechanistically, proteins in the mitochondrial respiratory chain complex and TCA cycle (NDUFS2, SDHA, ATP5B, and DLD) were the target of 4-HNE adduction in SOD2Δ hearts. Our findings suggest that the SOD2 mediated 4-HNE signaling nexus may play an important role in cardiomyopathy.
亲电醛(4-羟基壬烯醛;4-HNE)是脂质过氧化后形成的一种介质,它介导线粒体功能障碍,并与心血管疾病的发病机制和进展有关。锰超氧化物歧化酶(MnSOD)是一种核编码的抗氧化酶,它催化线粒体中超氧自由基(O)的歧化。为了研究 MnSOD 在心肌中的作用,我们生成了一种心肌细胞特异性 SOD2(SOD2Δ)缺陷的小鼠品系。与全身性 SOD2 敲除小鼠不同,SOD2Δ 小鼠可以进入青春期;然而,它们会在 4 个月左右的年龄因心力衰竭而死亡。SOD2Δ 心脏的超微结构分析显示线粒体结构发生改变,嵴和空泡形成明显破坏。SOD2 小鼠的非侵入性超声心动图测量显示扩张型心肌病特征,如射血分数和缩短分数降低,同时左心室内径增大。在 SOD2Δ 小鼠的心脏电生理研究中观察到室性心动过速的发生率增加。使用 Seahorse XF 分析仪对 SOD2Δ 新生心肌细胞和成年心脏线粒体进行的氧化磷酸化(OXPHOS)测量显示,与 SOD2fl 心脏相比,O 消耗减少,特别是在基础条件下和添加 FCCP(H 离子载体/解偶联剂)后。对这些细胞中糖酵解的细胞外酸化(ECAR)测量显示,与 SOD2 同窝仔鼠相比,SOD2Δ 小鼠的耗氧量(OCR)呈现出完全相反的模式。分析电子传递链复合物的活性表明,与 SOD2fl 小鼠相比,SOD2Δ 中的复合物 I 和复合物 V 活性降低。我们证明 SOD2 的缺乏会增加活性氧(ROS),导致线粒体中 4-HNE 的过度产生。从机制上讲,线粒体呼吸链复合物和 TCA 循环中的蛋白质(NDUFS2、SDHA、ATP5B 和 DLD)是 SOD2Δ 心脏中 4-HNE 加成的靶标。我们的研究结果表明,SOD2 介导的 4-HNE 信号连接可能在心肌病中发挥重要作用。