Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, International Centre for Life, Central Parkway, Newcastle upon Tyne, NE1 3BZ, UK.
Renal Services, The Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, NE7 7DN, UK.
BMC Nephrol. 2020 Oct 15;21(1):435. doi: 10.1186/s12882-020-02094-z.
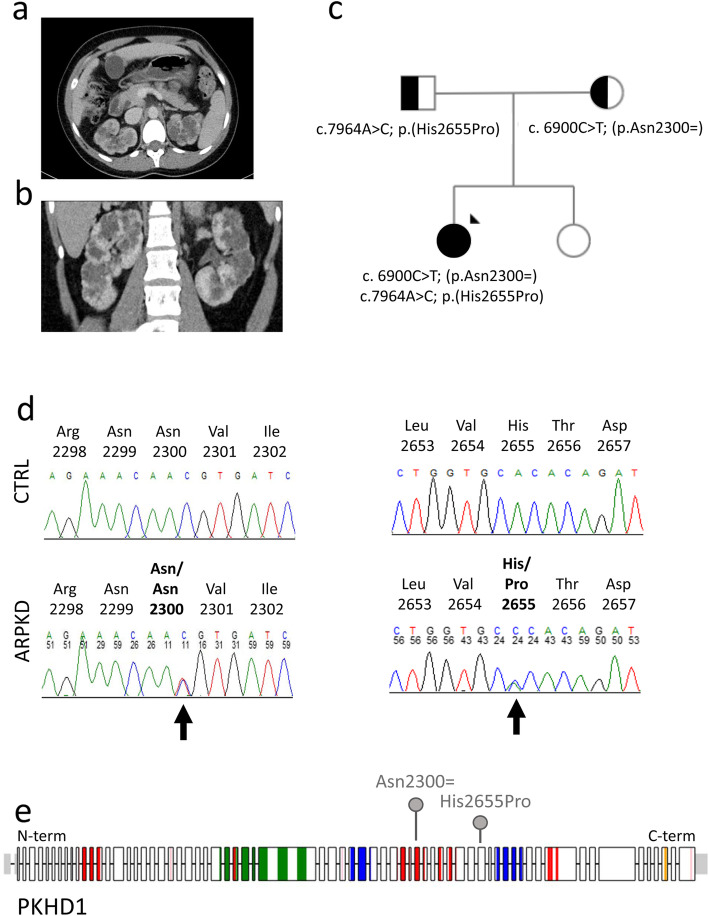
PKHD1 is the main genetic cause of autosomal recessive polycystic kidney disease (ARPKD), a hereditary hepato-renal fibrocystic disorder which is the most important cause of end-stage renal disease during early childhood. ARPKD can also present in adulthood with milder phenotypes. In this study, we describe a 24-year-old woman with atypical polycystic kidney, no family history of renal disease and no obvious extra-renal manifestations who was referred for genetic investigation.
We used a combination of next generation sequencing, Sanger sequencing and RNA and microscopy studies performed on urine-derived renal epithelial cells (URECs) to provide a genetic diagnosis of ARPKD.
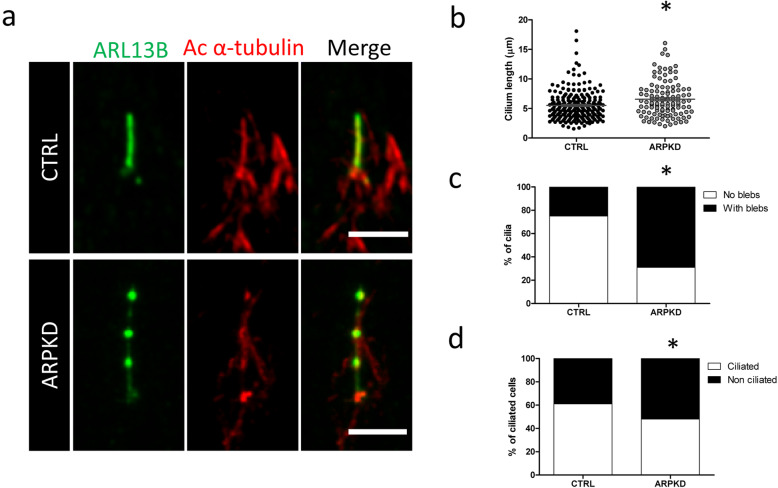
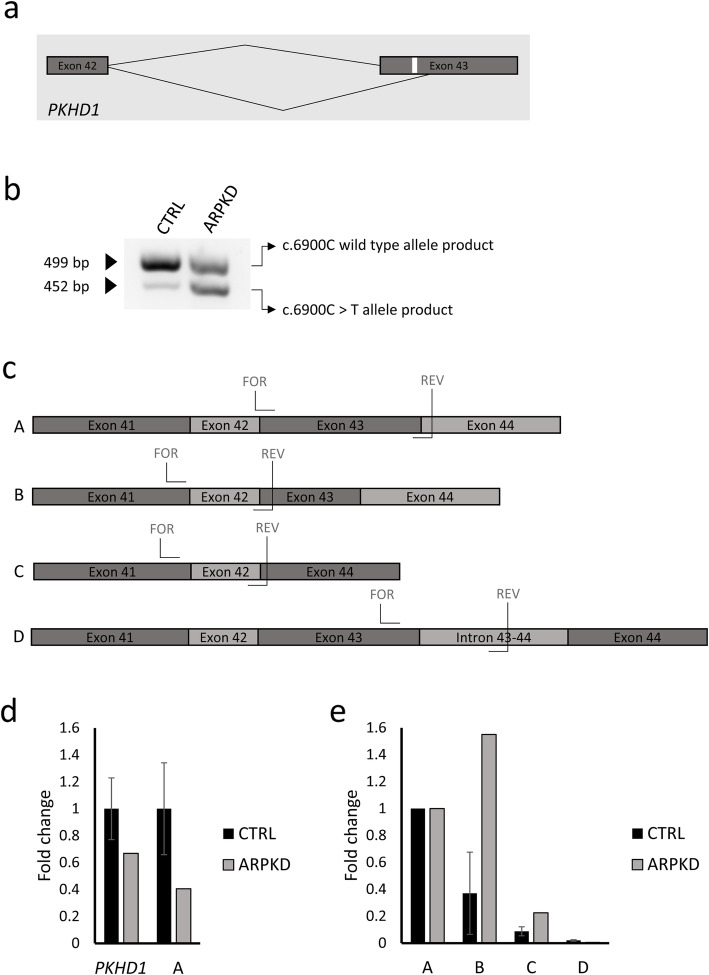
A next generation sequencing panel of cystic ciliopathy genes allowed the identification of two heterozygous sequence changes in PKHD1 (c.6900C > T; p.(Asn2300=) and c.7964A > C; p.(His2655Pro)). The pathogenicity of the synonymous PKHD1 variant is not clear and requires RNA studies, which cannot be carried out efficiently on RNA extracted from proband blood, due to the low expression levels of PKHD1 in lymphocytes. Using URECs as a source of kidney-specific RNA, we show that PKHD1 is alternatively spliced around exon 43, both in control and proband URECs. The variant p.(Asn2300=) shifts the expression ratio in favour of a shorter, out-of-frame transcript. To further study the phenotypic consequence of these variants, we investigated the ciliary phenotype of patient URECs, which were abnormally elongated and presented multiple blebs along the axoneme.
We confirm the power of URECs as a tool for functional studies on candidate variants in inherited renal disease, especially when the expression of the gene of interest is restricted to the kidney and we describe, for the first time, ciliary abnormalities in ARPKD patient cells.
PKHD1 是常染色体隐性多囊肾病(ARPKD)的主要遗传原因,ARPKD 是一种遗传性肝-肾纤维囊性疾病,是儿童早期终末期肾病的最重要原因。ARPKD 也可在成年期表现出较轻的表型。在这项研究中,我们描述了一位 24 岁的女性,其表现为非典型多囊肾,无肾脏疾病家族史,无明显的肾外表现,因遗传调查而被转介。
我们使用下一代测序、Sanger 测序以及尿液衍生的肾上皮细胞(UREC)中的 RNA 和显微镜研究的组合,提供了 ARPKD 的基因诊断。
囊性纤毛病基因的下一代测序面板允许鉴定 PKHD1 中的两个杂合序列变化(c.6900C>T;p.(Asn2300=)和 c.7964A>C;p.(His2655Pro))。同义 PKHD1 变体的致病性尚不清楚,需要 RNA 研究,但由于淋巴细胞中 PKHD1 的表达水平较低,无法有效地从先证者的血液中提取 RNA 进行 RNA 研究。使用 UREC 作为肾特异性 RNA 的来源,我们表明 PKHD1 在控制和先证者 UREC 中都在 43 号外显子周围发生选择性剪接。变体 p.(Asn2300=)改变了表达比例,有利于较短的、无框转录物。为了进一步研究这些变体的表型后果,我们研究了患者 UREC 的纤毛表型,发现其纤毛异常伸长,并在轴突上出现多个小泡。
我们证实了 UREC 作为研究遗传性肾脏疾病候选变异的功能研究工具的强大功能,特别是当感兴趣的基因的表达仅限于肾脏时,并且我们首次描述了 ARPKD 患者细胞中的纤毛异常。