Department of Nephrology, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka, 565-0871, Japan.
Department of Advanced Technology of Transplantation, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka, 565-0871, Japan.
Sci Rep. 2017 Aug 10;7(1):7733. doi: 10.1038/s41598-017-08284-4.
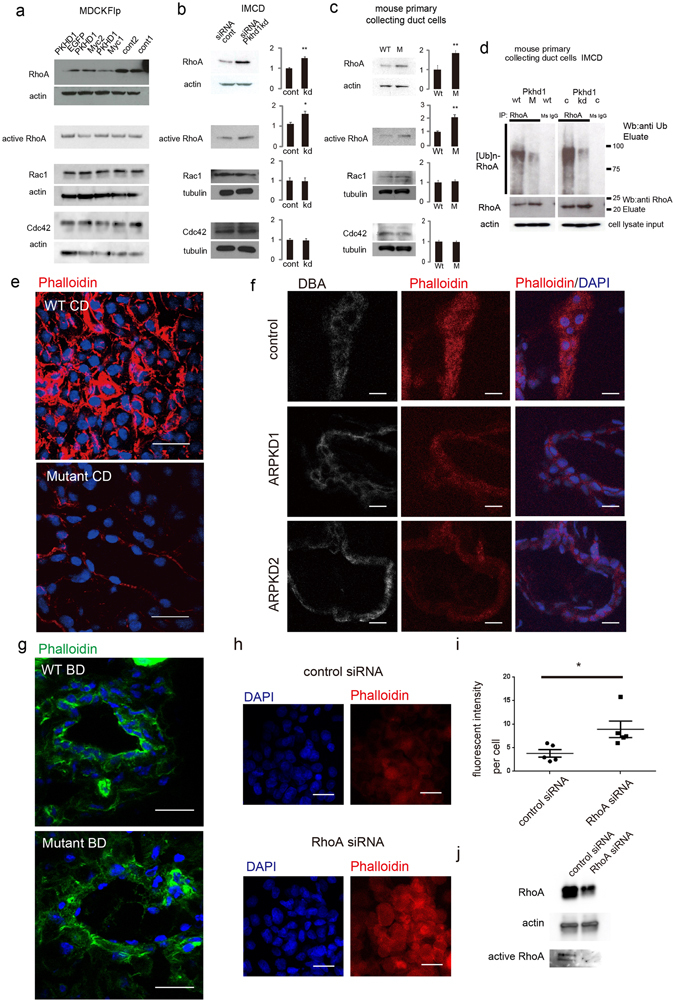
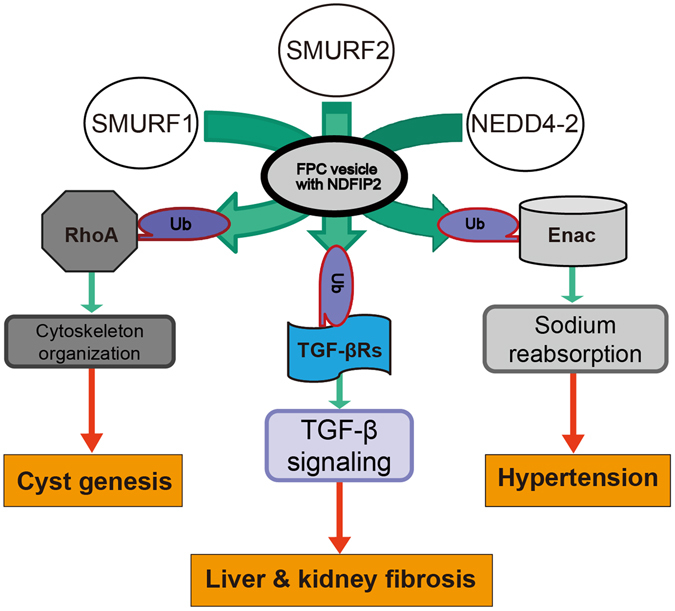
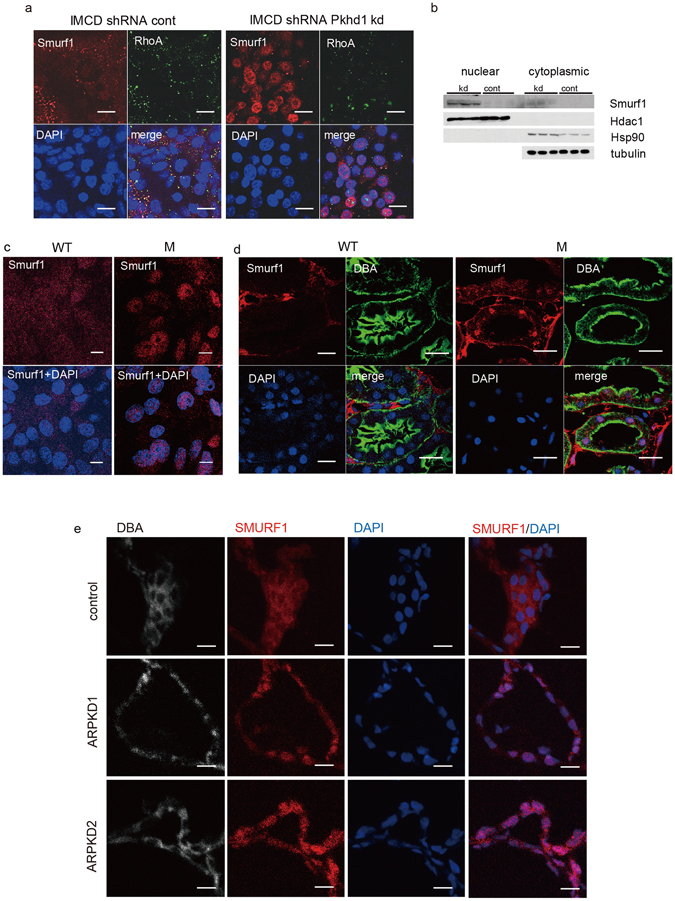
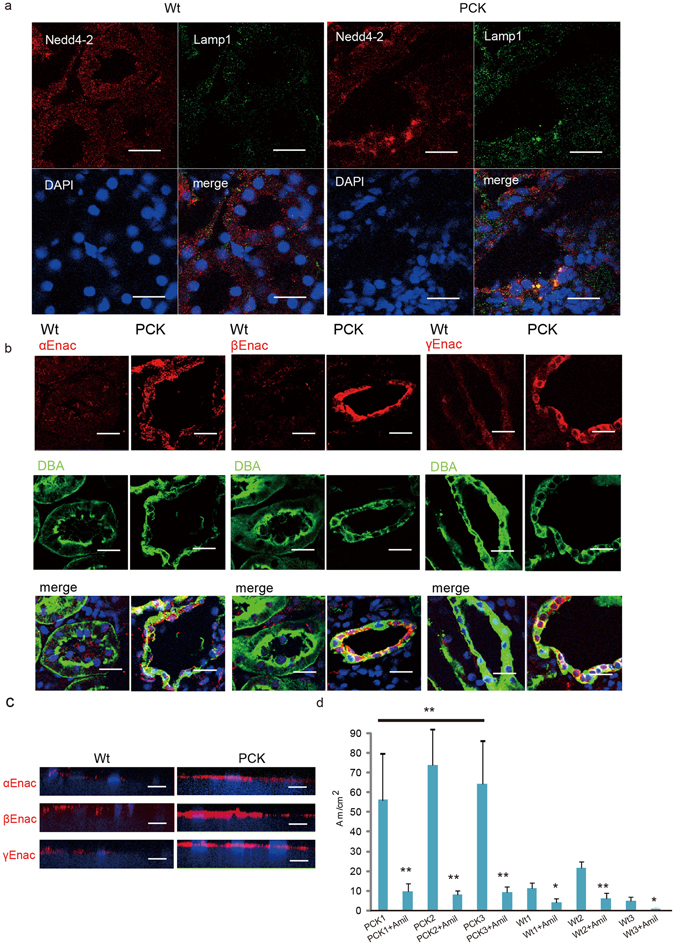
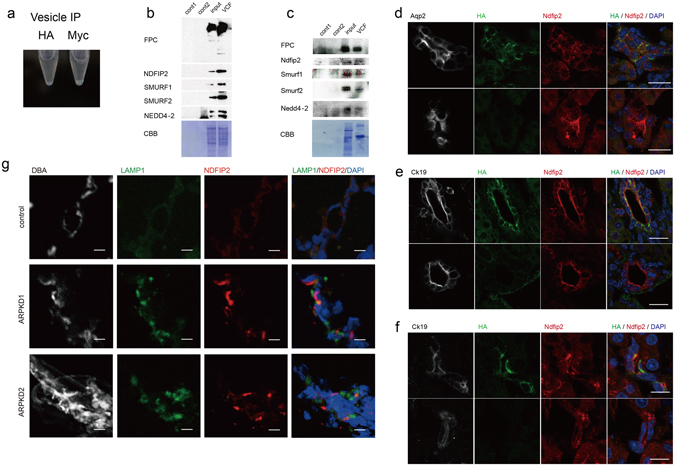
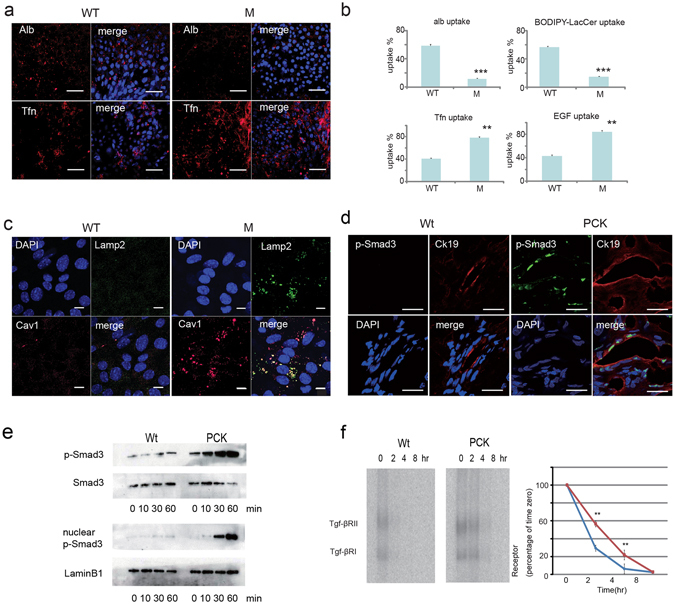
Autosomal recessive polycystic kidney disease (ARPKD) is an important childhood nephropathy, occurring 1 in 20,000 live births. The major clinical phenotypes are expressed in the kidney with dilatation of the collecting ducts, systemic hypertension, and progressive renal insufficiency, and in the liver with biliary dysgenesis, portal tract fibrosis, and portal hypertension. The systemic hypertension has been attributed to enhanced distal sodium reabsorption in the kidney, the structural defects have been ascribed to altered cellular morphology, and fibrosis to increased TGF-β signaling in the kidney and biliary tract, respectively. The pathogenic mechanisms underlying these abnormalities have not been determined. In the current report, we find that disrupting PKHD1 results in altered sub-cellular localization and function of the C2-WWW-HECT domain E3 family of ligases regulating these processes. We also demonstrate altered activity of RhoA and increased TGF-β signaling and ENaC activity. Linking these phenomena, we found that vesicles containing the PKHD1/Pkhd1 gene product, FPC, also contain the NEDD4 ubiquitin ligase interacting protein, NDFIP2, which interacts with multiple members of the C2-WWW-HECT domain E3 family of ligases. Our results provide a mechanistic explanation for both the cellular effects and in vivo phenotypic abnormalities in mice and humans that result from Pkhd1/PKHD1 mutation.
常染色体隐性多囊肾病(ARPKD)是一种重要的儿童肾病,每 20000 例活产中就有 1 例。主要的临床表型在肾脏表现为集合管扩张、全身高血压和进行性肾功能不全,在肝脏表现为胆管发育不良、门脉区纤维化和门静脉高压。全身高血压归因于肾脏中远端钠重吸收的增强,结构缺陷归因于细胞形态的改变,纤维化归因于肾脏和胆管中 TGF-β信号的增加。这些异常的发病机制尚未确定。在本报告中,我们发现破坏 PKHD1 导致调节这些过程的 C2-WWW-HECT 结构域 E3 家族连接酶的亚细胞定位和功能改变。我们还证明了 RhoA 活性的改变以及 TGF-β信号和 ENaC 活性的增加。将这些现象联系起来,我们发现含有 PKHD1/Pkhd1 基因产物 FPC 的囊泡也含有 NEDD4 泛素连接酶相互作用蛋白 NDFIP2,它与 C2-WWW-HECT 结构域 E3 家族的多个连接酶相互作用。我们的结果为 Pkhd1/PKHD1 突变导致的小鼠和人类的细胞效应和体内表型异常提供了一种机制解释。