The Province and Ministry Co-sponsored Collaborative Innovation Center for Medical Epigenetics, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin 300070, China.
Department of Pharmacology, Tianjin Key Laboratory of Inflammation Biology, School of Basic Medical Sciences, Tianjin Medical University, Tianjin 300070, China.
Genome Res. 2020 Dec;30(12):1789-1801. doi: 10.1101/gr.267997.120. Epub 2020 Oct 15.
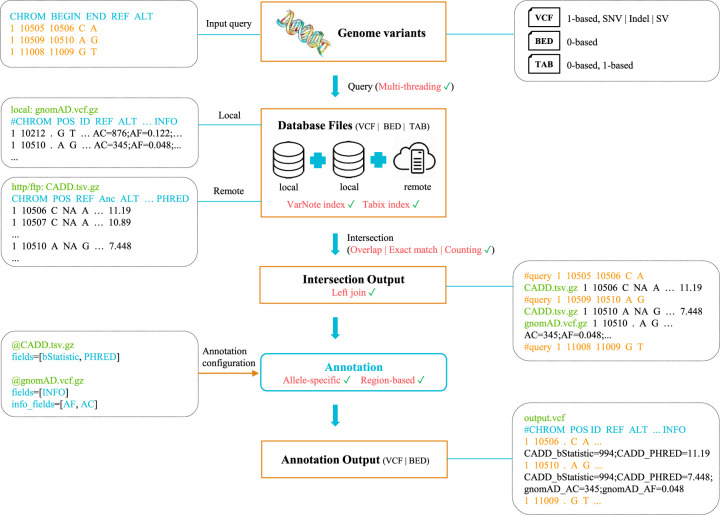
The advances of large-scale genomics studies have enabled compilation of cell type-specific, genome-wide DNA functional elements at high resolution. With the growing volume of functional annotation data and sequencing variants, existing variant annotation algorithms lack the efficiency and scalability to process big genomic data, particularly when annotating whole-genome sequencing variants against a huge database with billions of genomic features. Here, we develop VarNote to rapidly annotate genome-scale variants in large and complex functional annotation resources. Equipped with a novel index system and a parallel random-sweep searching algorithm, VarNote shows substantial performance improvements (two to three orders of magnitude) over existing algorithms at different scales. It supports both region-based and allele-specific annotations and introduces advanced functions for the flexible extraction of annotations. By integrating massive base-wise and context-dependent annotations in the VarNote framework, we introduce three efficient and accurate pipelines to prioritize the causal regulatory variants for common diseases, Mendelian disorders, and cancers.
大规模基因组学研究的进展使得能够以高分辨率编译细胞类型特异性的全基因组 DNA 功能元件。随着功能注释数据和测序变体数量的不断增加,现有的变体注释算法在处理大型基因组数据时效率和可扩展性不足,特别是在针对具有数十亿个基因组特征的庞大数据库注释全基因组测序变体时。在这里,我们开发了 VarNote 来快速注释大型复杂功能注释资源中的基因组规模变体。VarNote 配备了新颖的索引系统和并行随机扫描搜索算法,在不同规模上相对于现有算法具有显著的性能提升(两个到三个数量级)。它支持基于区域和等位基因特异性的注释,并引入了用于灵活提取注释的高级功能。通过在 VarNote 框架中集成大量基于碱基和上下文相关的注释,我们引入了三种高效准确的管道,用于为常见疾病、孟德尔疾病和癌症优先排序因果调控变体。