Jia Rong, Li Zhongxian, Liang Wei, Ji Yucheng, Weng Yujie, Liang Ying, Ning Pengfei
College of Computer and Information, Inner Mongolia Medical University, Hohhot, 010110, Inner Mongolia Autonomous Region, China.
World J Surg Oncol. 2020 Oct 16;18(1):268. doi: 10.1186/s12957-020-02042-z.
Breast cancer subtypes are statistically associated with prognosis. The search for markers of breast tumor heterogeneity and the development of precision medicine for patients are the current focuses of the field.
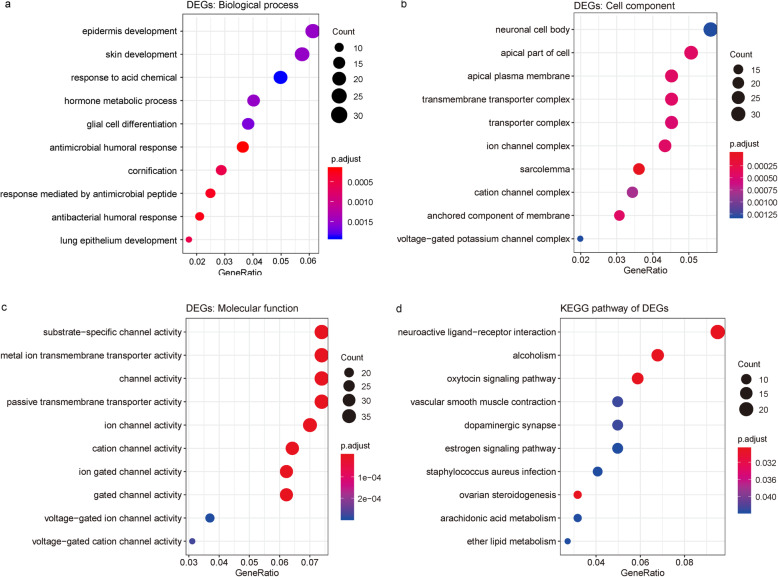
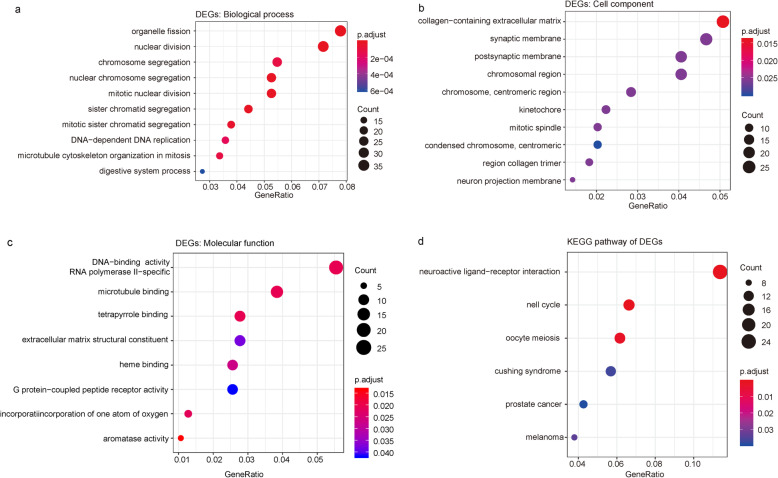
We used a bioinformatic approach to identify key disease-causing genes unique to the luminal A and basal-like subtypes of breast cancer. First, we retrieved gene expression data for luminal A breast cancer, basal-like breast cancer, and normal breast tissue samples from The Cancer Genome Atlas database. The differentially expressed genes unique to the 2 breast cancer subtypes were identified and subjected to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses. We constructed protein-protein interaction networks of the differentially expressed genes. Finally, we analyzed the key modules of the networks, which we combined with survival data to identify the unique cancer genes associated with each breast cancer subtype.
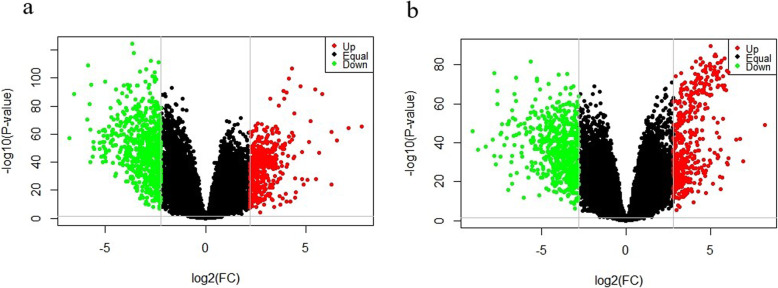
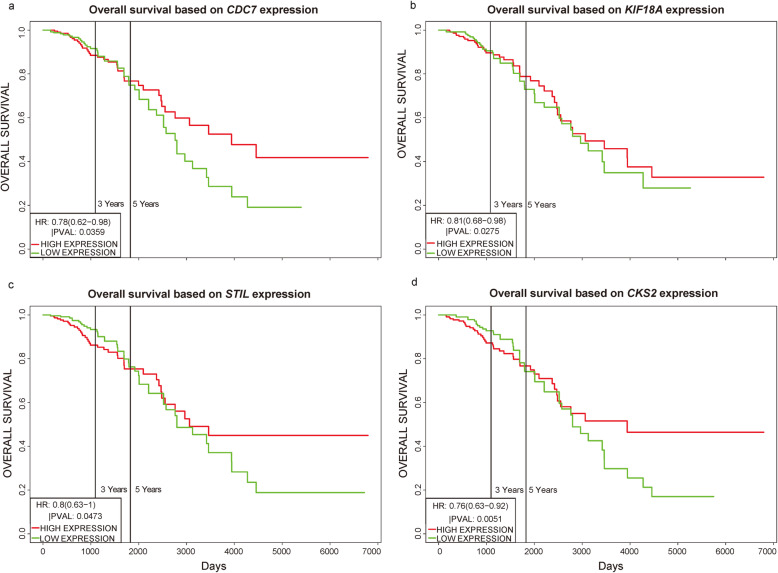
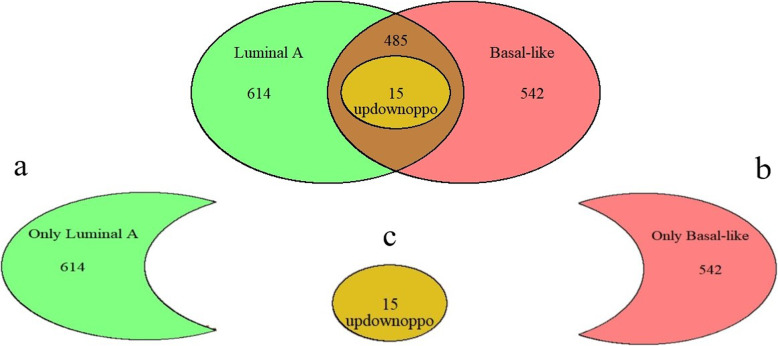
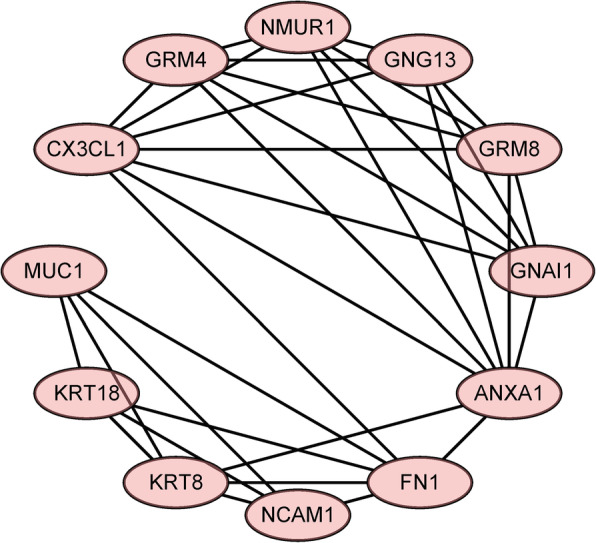
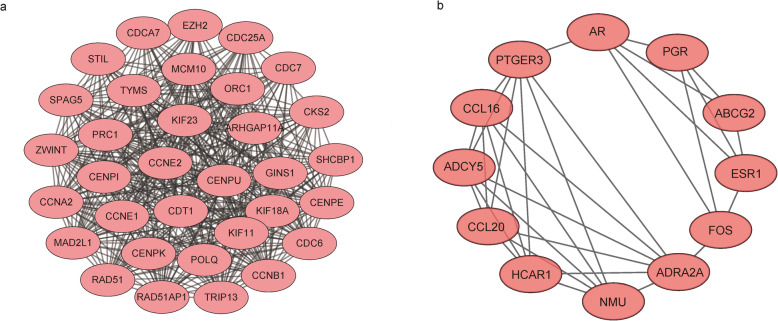
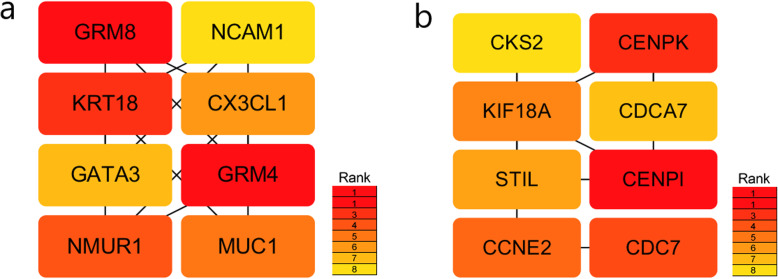
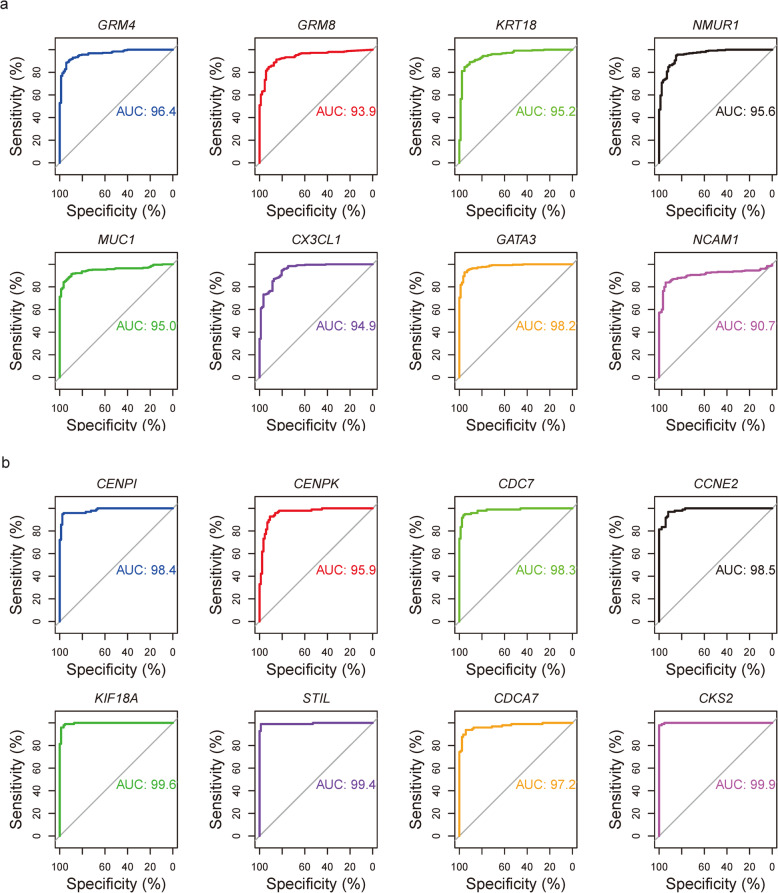
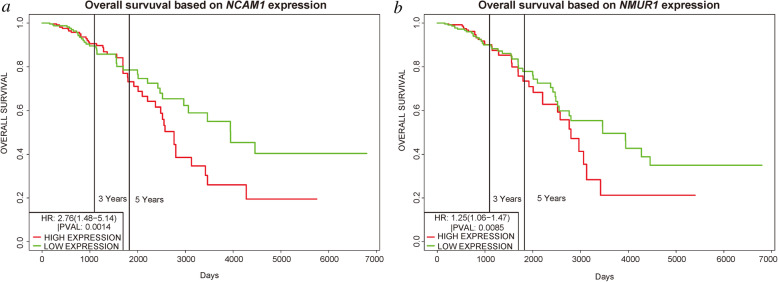
We identified 1114 differentially expressed genes in luminal A breast cancer and 1042 differentially expressed genes in basal-like breast cancer, of which the subtypes shared 500. We observed 614 and 542 differentially expressed genes unique to luminal A and basal-like breast cancer, respectively. Through enrichment analyses, protein-protein interaction network analysis, and module mining, we identified 8 key differentially expressed genes unique to each subtype. Analysis of the gene expression data in the context of the survival data revealed that high expression of NMUR1 and NCAM1 in luminal A breast cancer statistically correlated with poor prognosis, whereas the low expression levels of CDC7, KIF18A, STIL, and CKS2 in basal-like breast cancer statistically correlated with poor prognosis.
NMUR1 and NCAM1 are novel key disease-causing genes for luminal A breast cancer, and STIL is a novel key disease-causing gene for basal-like breast cancer. These genes are potential targets for clinical treatment.
乳腺癌亚型与预后在统计学上相关。寻找乳腺肿瘤异质性的标志物以及为患者开发精准医学是该领域当前的重点。
我们采用生物信息学方法来鉴定乳腺腔面A型和基底样亚型特有的关键致病基因。首先,我们从癌症基因组图谱数据库中检索了乳腺腔面A型乳腺癌、基底样乳腺癌和正常乳腺组织样本的基因表达数据。鉴定出这两种乳腺癌亚型特有的差异表达基因,并对其进行基因本体论和京都基因与基因组百科全书通路富集分析。我们构建了差异表达基因的蛋白质-蛋白质相互作用网络。最后,我们分析了网络的关键模块,并将其与生存数据相结合,以鉴定与每种乳腺癌亚型相关的独特癌症基因。
我们在乳腺腔面A型乳腺癌中鉴定出1114个差异表达基因,在基底样乳腺癌中鉴定出1042个差异表达基因,其中两种亚型共有500个。我们分别观察到乳腺腔面A型和基底样乳腺癌特有的614个和542个差异表达基因。通过富集分析、蛋白质-蛋白质相互作用网络分析和模块挖掘,我们鉴定出每种亚型特有的8个关键差异表达基因。在生存数据背景下对基因表达数据的分析表明,NMUR1和NCAM1在乳腺腔面A型乳腺癌中的高表达与预后不良在统计学上相关,而CDC7、KIF18A、STIL和CKS2在基底样乳腺癌中的低表达与预后不良在统计学上相关。
NMUR1和NCAM1是乳腺腔面A型乳腺癌新的关键致病基因,STIL是基底样乳腺癌新的关键致病基因。这些基因是临床治疗的潜在靶点。