LungenClinic Grosshansdorf, Großhansdorf, Germany.
Airway Research Center North (ARCN), Member of the German Center for Lung Research (DZL), Großhansdorf, Germany.
Respir Res. 2020 Oct 19;21(1):274. doi: 10.1186/s12931-020-01544-4.
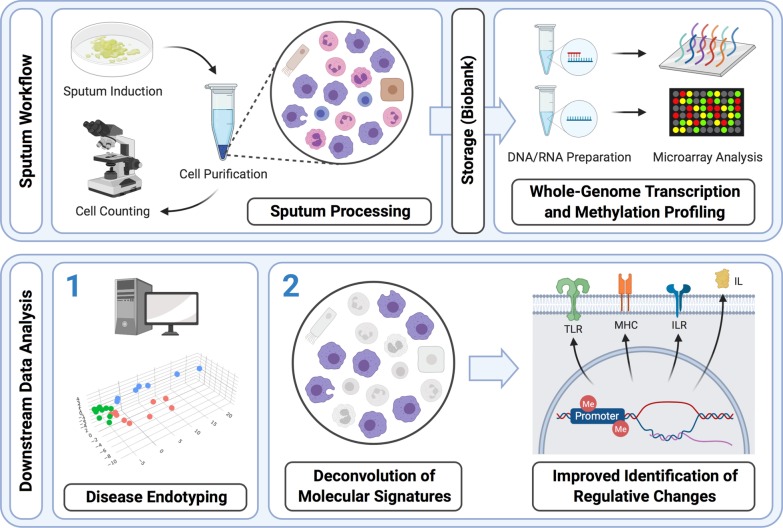
To date, most studies involving high-throughput analyses of sputum in asthma and COPD have focused on identifying transcriptomic signatures of disease. No whole-genome methylation analysis of sputum cells has been performed yet. In this context, the highly variable cellular composition of sputum has potential to confound the molecular analyses.
Whole-genome transcription (Agilent Human 4 × 44 k array) and methylation (Illumina 450 k BeadChip) analyses were performed on sputum samples of 9 asthmatics, 10 healthy and 10 COPD subjects. RNA integrity was checked by capillary electrophoresis and used to correct in silico for bias conferred by RNA degradation during biobank sample storage. Estimates of cell type-specific molecular profiles were derived via regression by quadratic programming based on sputum differential cell counts. All analyses were conducted using the open-source R/Bioconductor software framework.
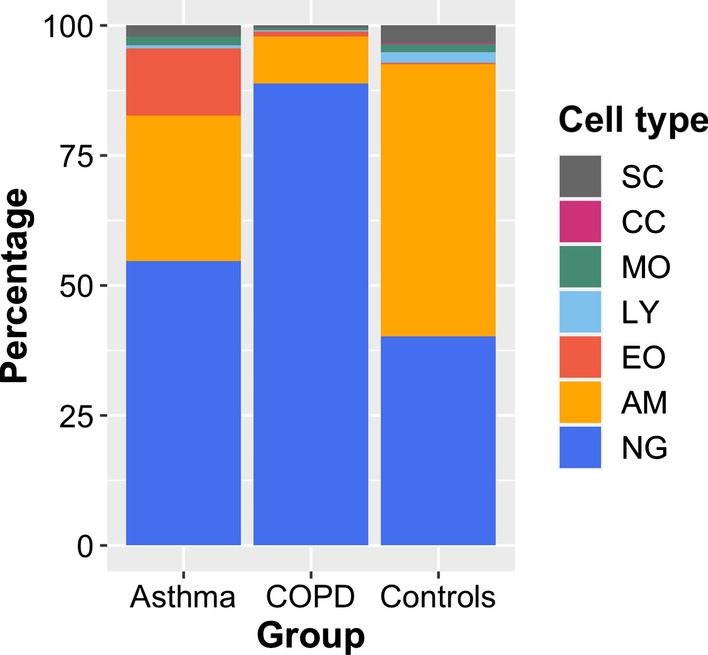
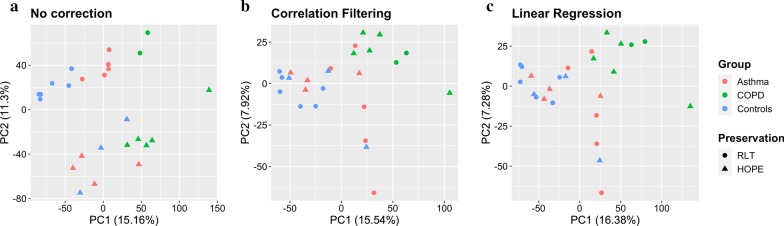
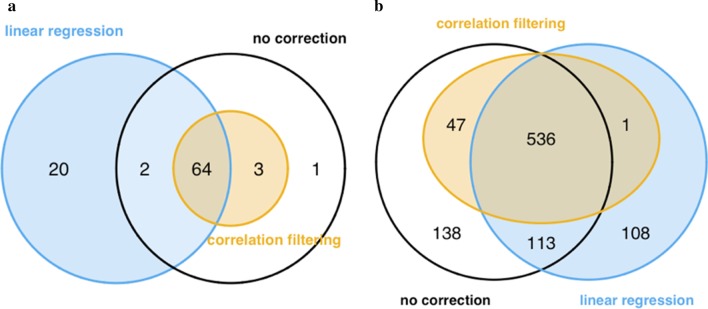
A linear regression step was found to perform well in removing RNA degradation-related bias among the main principal components of the gene expression data, increasing the number of genes detectable as differentially expressed in asthma and COPD sputa (compared to controls). We observed a strong influence of the cellular composition on the results of mixed-cell sputum analyses. Exemplarily, upregulated genes derived from mixed-cell data in asthma were dominated by genes predominantly expressed in eosinophils after deconvolution. The deconvolution, however, allowed to perform differential expression and methylation analyses on the level of individual cell types and, though we only analyzed a limited number of biological replicates, was found to provide good estimates compared to previously published data about gene expression in lung eosinophils in asthma. Analysis of the sputum methylome indicated presence of differential methylation in genomic regions of interest, e.g. mapping to a number of human leukocyte antigen (HLA) genes related to both major histocompatibility complex (MHC) class I and II molecules in asthma and COPD macrophages. Furthermore, we found the SMAD3 (SMAD family member 3) gene, among others, to lie within differentially methylated regions which has been previously reported in the context of asthma.
In this methodology-oriented study, we show that methylation profiling can be easily integrated into sputum analysis workflows and exhibits a strong potential to contribute to the profiling and understanding of pulmonary inflammation. Wherever RNA degradation is of concern, in silico correction can be effective in improving both sensitivity and specificity of downstream analyses. We suggest that deconvolution methods should be integrated in sputum omics analysis workflows whenever possible in order to facilitate the unbiased discovery and interpretation of molecular patterns of inflammation.
迄今为止,大多数涉及哮喘和 COPD 患者痰液高通量分析的研究都集中在鉴定疾病的转录组特征上。尚未对痰液细胞进行全基因组甲基化分析。在这种情况下,痰液高度可变的细胞组成可能会混淆分子分析。
对 9 名哮喘患者、10 名健康对照者和 10 名 COPD 患者的痰液样本进行全基因组转录(安捷伦人类 4×44k 芯片)和甲基化(Illumina 450k BeadChip)分析。通过毛细管电泳检查 RNA 完整性,并在生物库样本储存过程中使用 RNA 降解导致的偏倚的校正。通过基于痰液差异细胞计数的二次规划回归来估计细胞类型特异性分子谱的估计值。所有分析均使用开源 R/Bioconductor 软件框架进行。
线性回归步骤在去除基因表达数据主要主成分中与 RNA 降解相关的偏倚方面表现良好,增加了在哮喘和 COPD 痰液中可检测到差异表达的基因数量(与对照相比)。我们观察到细胞组成对混合细胞痰液分析结果的强烈影响。例如,在哮喘中从混合细胞数据中推导出来的上调基因主要由嗜酸性粒细胞中表达的基因主导。然而,去卷积允许在单个细胞类型水平上进行差异表达和甲基化分析,并且尽管我们只分析了有限数量的生物学重复,但与之前关于哮喘中肺嗜酸性粒细胞的基因表达的发表数据相比,发现它提供了很好的估计。痰液甲基化组分析表明,在感兴趣的基因组区域存在差异甲基化,例如哮喘和 COPD 巨噬细胞中与主要组织相容性复合物(MHC)I 类和 II 类分子相关的多个人类白细胞抗原(HLA)基因。此外,我们发现 SMAD3(SMAD 家族成员 3)基因等位于先前在哮喘背景下报道的差异甲基化区域内。
在这项面向方法的研究中,我们表明甲基化分析可以很容易地整合到痰液分析工作流程中,并具有为肺部炎症的分析和理解提供有力支持的巨大潜力。只要存在 RNA 降解的问题,计算机校正就可以有效地提高下游分析的敏感性和特异性。我们建议,只要有可能,应将去卷积方法集成到痰液组学分析工作流程中,以便于发现和解释炎症的分子模式。