Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia.
Department of Medical Biology, The University of Melbourne, Melbourne, VIC, Australia.
Malar J. 2020 Oct 20;19(1):375. doi: 10.1186/s12936-020-03440-0.
Genomic surveillance of malaria parasite populations has the potential to inform control strategies and to monitor the impact of interventions. Barcodes comprising large numbers of single nucleotide polymorphism (SNP) markers are accurate and efficient genotyping tools, however may need to be tailored to specific malaria transmission settings, since 'universal' barcodes can lack resolution at the local scale. A SNP barcode was developed that captures the diversity and structure of Plasmodium vivax populations of Papua New Guinea (PNG) for research and surveillance.
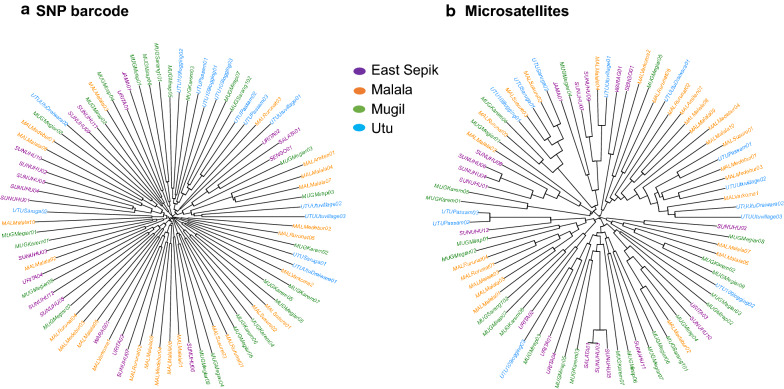
Using 20 high-quality P. vivax genome sequences from PNG, a total of 178 evenly spaced neutral SNPs were selected for development of an amplicon sequencing assay combining a series of multiplex PCRs and sequencing on the Illumina MiSeq platform. For initial testing, 20 SNPs were amplified in a small number of mono- and polyclonal P. vivax infections. The full barcode was then validated by genotyping and population genetic analyses of 94 P. vivax isolates collected between 2012 and 2014 from four distinct catchment areas on the highly endemic north coast of PNG. Diversity and population structure determined from the SNP barcode data was then benchmarked against that of ten microsatellite markers used in previous population genetics studies.
From a total of 28,934,460 reads generated from the MiSeq Illumina run, 87% mapped to the PvSalI reference genome with deep coverage (median = 563, range 56-7586) per locus across genotyped samples. Of 178 SNPs assayed, 146 produced high-quality genotypes (minimum coverage = 56X) in more than 85% of P. vivax isolates. No amplification bias was introduced due to either polyclonal infection or whole genome amplification (WGA) of samples before genotyping. Compared to the microsatellite panels, the SNP barcode revealed greater variability in genetic diversity between populations and geographical population structure. The SNP barcode also enabled assignment of genotypes according to their geographic origins with a significant association between genetic distance and geographic distance at the sub-provincial level.
High-throughput SNP barcoding can be used to map variation of malaria transmission dynamics at sub-national resolution. The low cost per sample and genotyping strategy makes the transfer of this technology to field settings highly feasible.
疟疾寄生虫种群的基因组监测具有为控制策略提供信息和监测干预措施影响的潜力。由大量单核苷酸多态性(SNP)标记组成的条码是准确和高效的基因分型工具,但可能需要根据特定的疟疾传播情况进行调整,因为“通用”条码在局部尺度上可能缺乏分辨率。开发了一种 SNP 条码,用于捕获巴布亚新几内亚(PNG)间日疟原虫种群的多样性和结构,用于研究和监测。
利用来自 PNG 的 20 个高质量间日疟原虫基因组序列,共选择了 178 个均匀间隔的中性 SNP,用于开发一种扩增子测序分析,该分析结合了一系列多重 PCR 和 Illumina MiSeq 平台上的测序。在少数单克隆和多克隆间日疟原虫感染中,首先对 20 个 SNP 进行了扩增。然后通过对 2012 年至 2014 年间在 PNG 高度流行的北海岸四个不同集水区采集的 94 个间日疟原虫分离株进行基因分型和种群遗传分析,对全条码进行了验证。然后将从 SNP 条码数据确定的多样性和种群结构与之前在种群遗传学研究中使用的 10 个微卫星标记进行了比较。
从 MiSeq Illumina 运行生成的总共 28,934,460 个读取中,87%的读取映射到 PvSalI 参考基因组,每个基因座的深度覆盖中位数为 563(范围为 56-7586),覆盖了所有基因分型样本。在检测的 178 个 SNP 中,146 个 SNP 在超过 85%的间日疟原虫分离株中产生了高质量的基因型(最小覆盖度为 56X)。由于多克隆感染或样本全基因组扩增(WGA),在基因分型之前没有引入扩增偏倚。与微卫星面板相比,SNP 条码揭示了种群之间和地理种群结构中遗传多样性的更大变异性。SNP 条码还可以根据其地理起源对基因型进行分配,并且在省级以下水平上,遗传距离与地理距离之间存在显著关联。
高通量 SNP 条码可用于以次国家分辨率绘制疟疾传播动态的变化。每个样本的低成本和基因分型策略使得将这项技术转移到现场设置非常可行。