Jayasheela K, Nagabalasubramanian P B, Periandy S
Department of Physics, Kanchi Mamunivar Centre for Postgraduate Studies, Puducherry, India.
Department of Physics, Arignar Anna Govt. Arts & Science College, Karaikal, Puducherry, India.
Heliyon. 2020 Oct 8;6(10):e04775. doi: 10.1016/j.heliyon.2020.e04775. eCollection 2020 Oct.
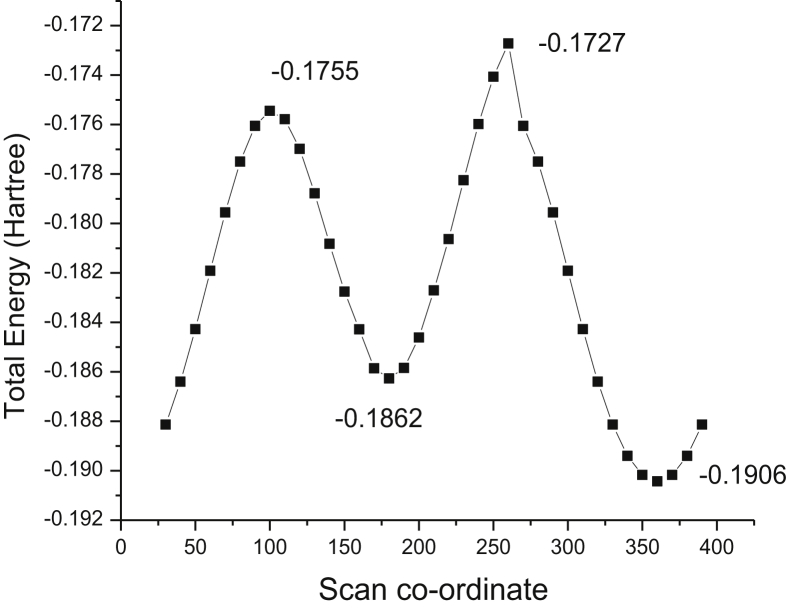
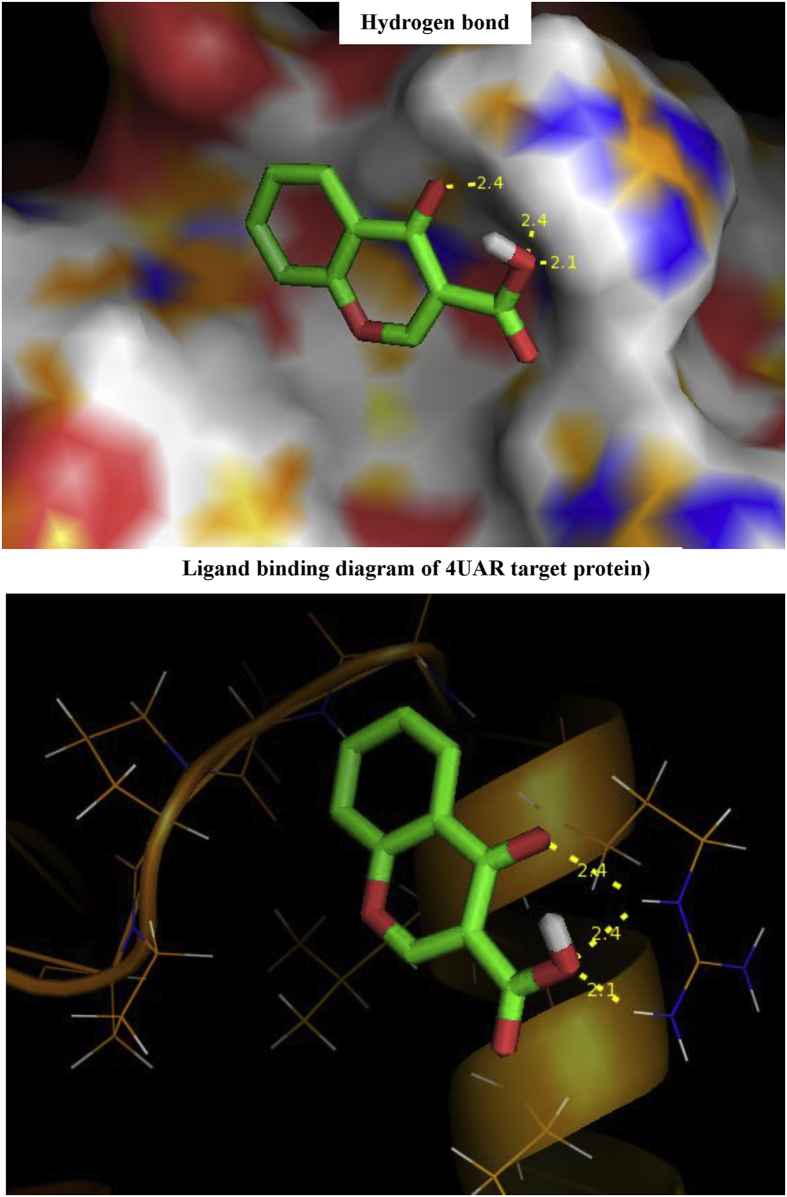
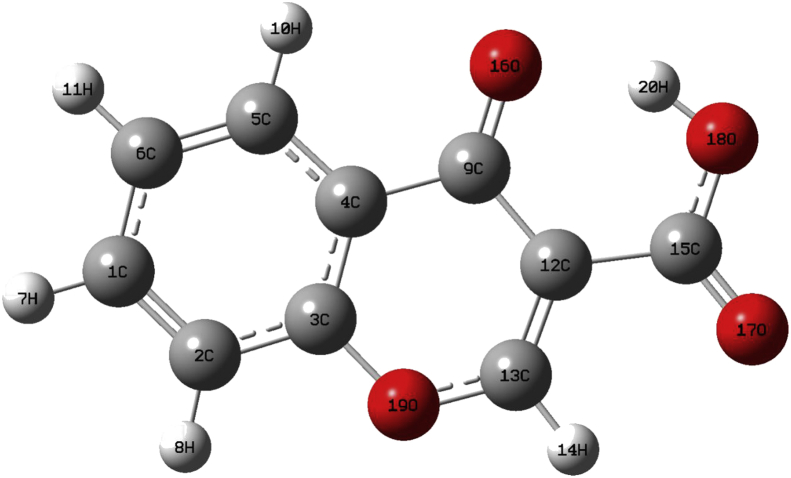
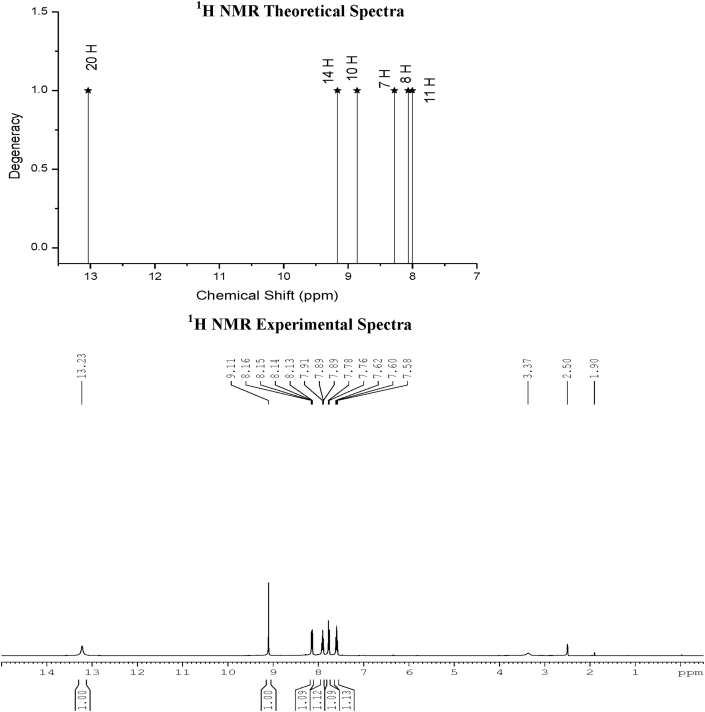
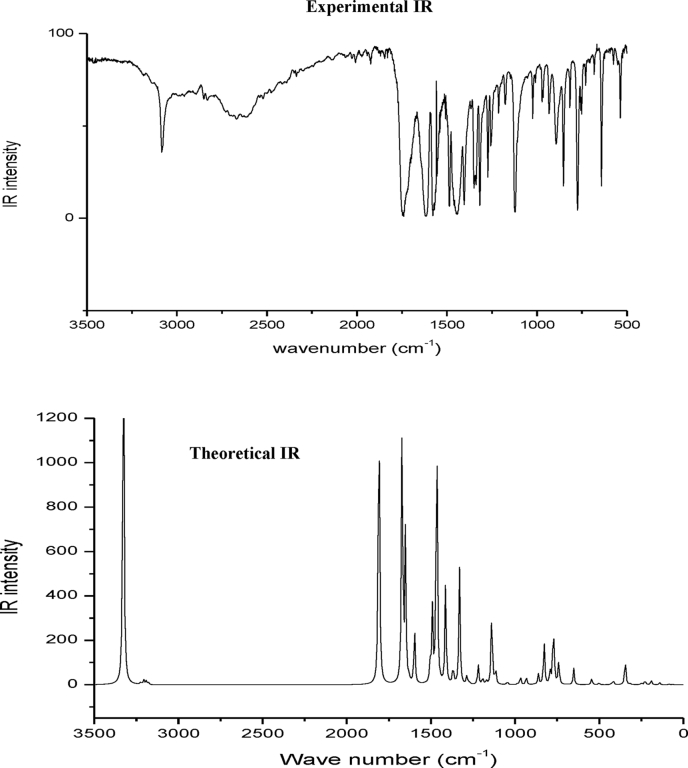
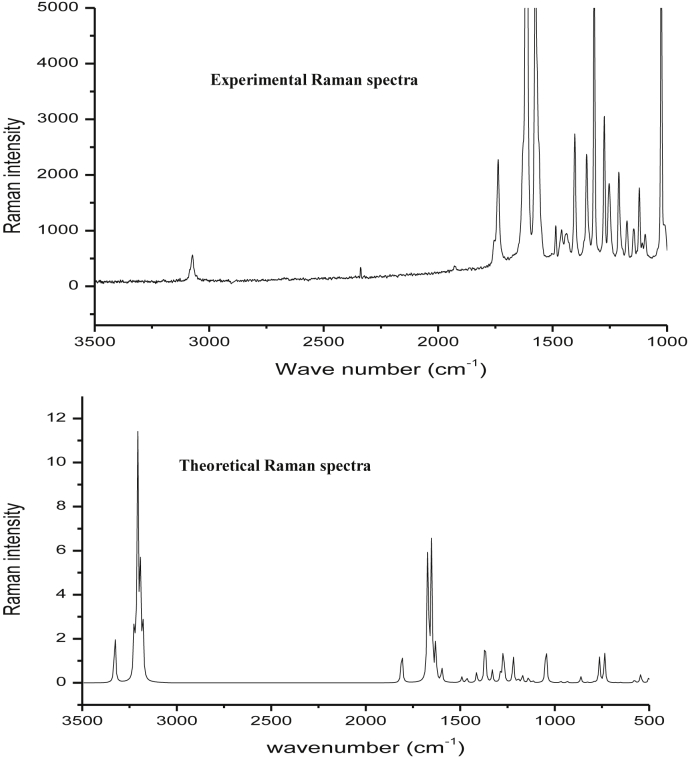
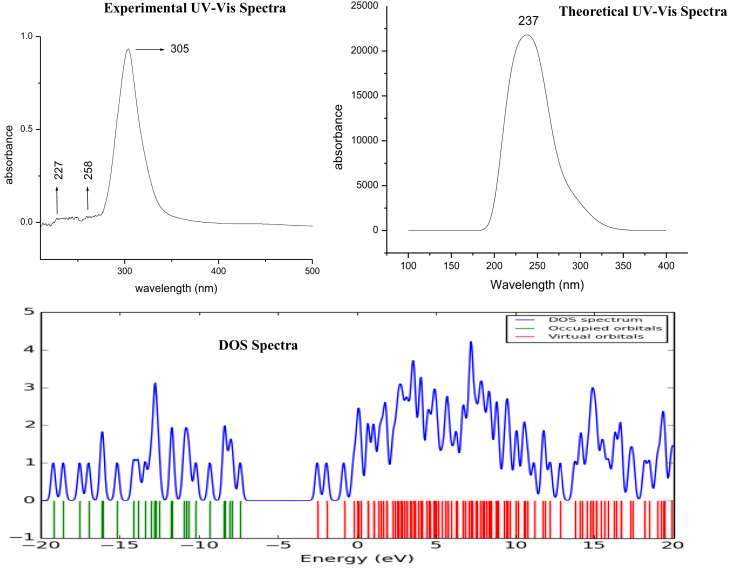
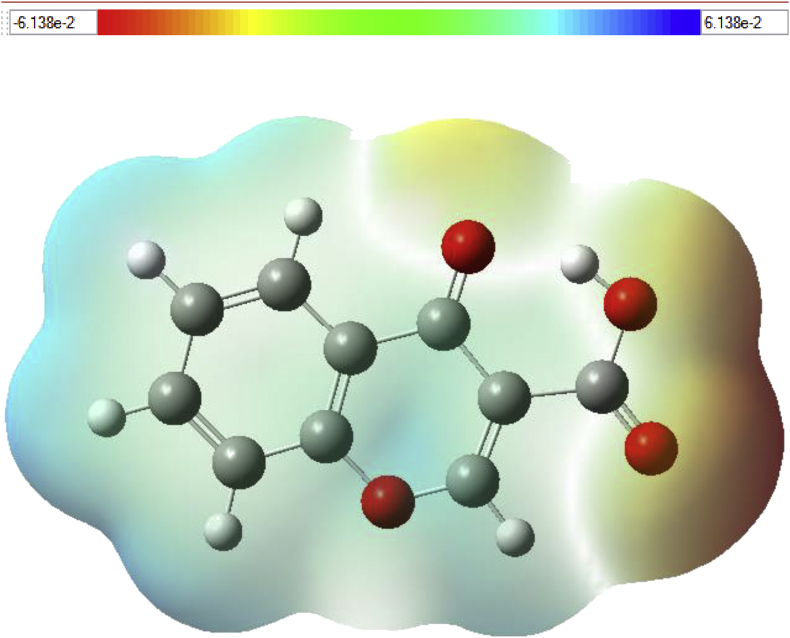
The Spectroscopic profile of Chromone-3-Carboxylic Acid (abbreviated as C3CA) was examined using FT-IR, FT-Raman, UV, H and C NMR techniques. The geometrical parameters and energies attained from DFT/B3LYP method with 6-311++G (d,p) basis sets calculations. The geometry of the molecule was fully optimized, vibrational spectra were calculated and assigned the fundamental vibrations on the basis of the total energy distribution (TED) of the vibrational modes, calculated with scaled quantum mechanics (SQM) method. The XRD data obtained from the computed geometric parameters shows that there is little deviation in the structure due to the substitution of the COOH group in the molecule. Using NBO study, the delocalization of the electron and the corresponding attraction between the orbitals shows that the lone pair transition has higher stabilization energy when compared with remaining atoms. The H and C NMR chemical shifts are calculated using GIAO method and the experimental chemical shifts were analysed with theoretical values which reflects better coincidence. The electronic properties, HOMO and LUMO energies, are performed with TD-DFT reproduces good with the experimental findings. Besides, frontier molecular orbitals (FMO), the high reactive nature of the molecule is identified with MEP and global reactivity descriptor analysis are performed. In addition, the molecular docking study was conducted, and results of the docking study identified the sugar phosphatase inhibitor activity of the target molecule (C3CA).
使用傅里叶变换红外光谱(FT-IR)、傅里叶变换拉曼光谱(FT-Raman)、紫外光谱(UV)、氢核磁共振(H NMR)和碳核磁共振(C NMR)技术研究了色酮-3-羧酸(简称为C3CA)的光谱特征。通过密度泛函理论(DFT)/B3LYP方法,采用6-311++G(d,p)基组计算获得了几何参数和能量。对分子的几何结构进行了完全优化,计算了振动光谱,并根据振动模式的总能量分布(TED),采用缩放量子力学(SQM)方法对基本振动进行了归属。从计算得到的几何参数获得的X射线衍射(XRD)数据表明,由于分子中COOH基团的取代,结构上存在微小偏差。通过自然键轨道(NBO)研究,电子的离域以及轨道之间的相应吸引力表明,与其余原子相比,孤对电子跃迁具有更高的稳定化能。使用GIAO方法计算了氢核磁共振(H NMR)和碳核磁共振(C NMR)化学位移,并将实验化学位移与理论值进行了分析,两者吻合较好。采用含时密度泛函理论(TD-DFT)计算了电子性质、最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)能量,与实验结果吻合良好。此外,通过前线分子轨道(FMO)、分子静电势(MEP)和全局反应活性描述符分析确定了该分子的高反应活性。此外,还进行了分子对接研究,对接研究结果确定了目标分子(C3CA)的糖磷酸酶抑制活性。