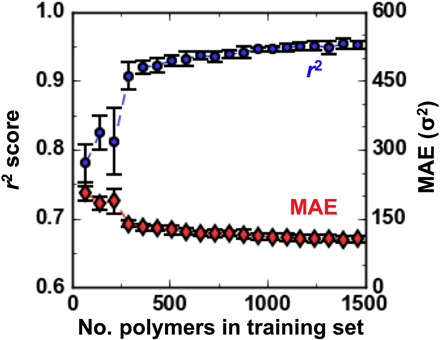
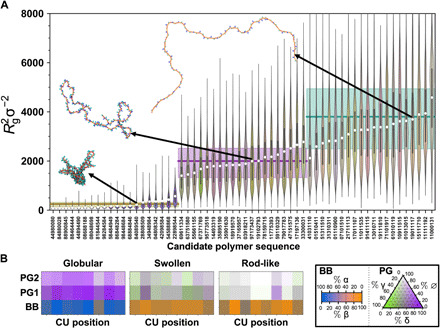
Webb Michael A, Jackson Nicholas E, Gil Phwey S, de Pablo Juan J
Pritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60615, USA.
Center for Molecular Engineering and Materials Science Division, Argonne National Laboratory, Lemont, IL 06349, USA.
Sci Adv. 2020 Oct 21;6(43). doi: 10.1126/sciadv.abc6216. Print 2020 Oct.
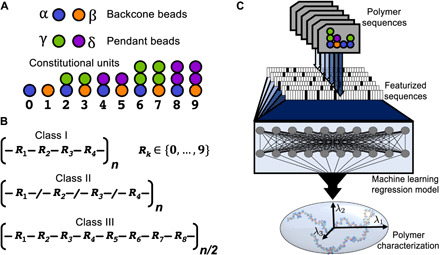
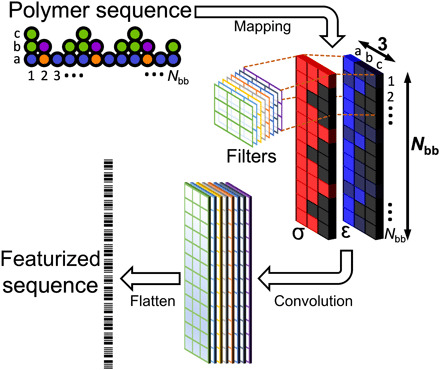
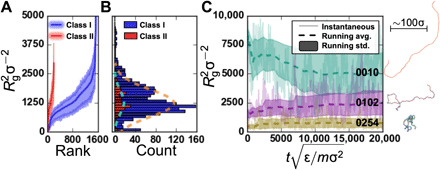
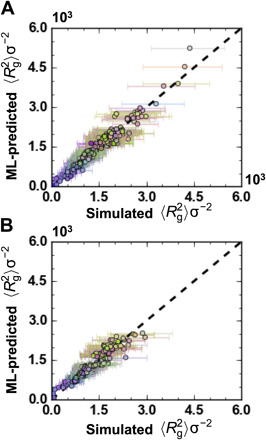
The chemical design of polymers with target structural and/or functional properties represents a grand challenge in materials science. While data-driven design approaches are promising, success with polymers has been limited, largely due to limitations in data availability. Here, we demonstrate the targeted sequence design of single-chain structure in polymers by combining coarse-grained modeling, machine learning, and model optimization. Nearly 2000 unique coarse-grained polymers are simulated to construct and analyze machine learning models. We find that deep neural networks inexpensively and reliably predict structural properties with limited sequence information as input. By coupling trained ML models with sequential model-based optimization, polymer sequences are proposed to exhibit globular, swollen, or rod-like behaviors, which are verified by explicit simulations. This work highlights the promising integration of coarse-grained modeling with data-driven design and represents a necessary and crucial step toward more complex polymer design efforts.
具有目标结构和/或功能特性的聚合物的化学设计是材料科学中的一项重大挑战。虽然数据驱动的设计方法很有前景,但聚合物方面的成功一直有限,这主要是由于数据可用性的限制。在这里,我们通过结合粗粒度建模、机器学习和模型优化,展示了聚合物中单链结构的靶向序列设计。模拟了近2000种独特的粗粒度聚合物,以构建和分析机器学习模型。我们发现,深度神经网络以有限的序列信息作为输入,可以廉价且可靠地预测结构特性。通过将训练好的机器学习模型与基于序列模型的优化相结合,提出了具有球状、膨胀状或棒状行为的聚合物序列,并通过显式模拟进行了验证。这项工作突出了粗粒度建模与数据驱动设计的有前景的整合,代表了朝着更复杂的聚合物设计努力迈出的必要且关键的一步。