Gierhardt Mareike, Pak Oleg, Sydykov Akylbek, Kraut Simone, Schäffer Julia, Garcia Claudia, Veith Christine, Zeidan Esraa M, Brosien Monika, Quanz Karin, Esfandiary Azadeh, Saraji Alireza, Hadzic Stefan, Kojonazarov Baktybek, Wilhelm Jochen, Ghofrani Hossein A, Schermuly Ralph T, Seeger Werner, Grimminger Friedrich, Herden Christiane, Schulz Rainer, Weissmann Norbert, Heger Jacqueline, Sommer Natascha
Excellence Cluster Cardio Pulmonary Institute (CPI), University of Giessen and Marburg Lung Center (UGMLC), Member of the German Center for Lung Research (DZL), Justus-Liebig University, Giessen, Germany.
Instituto de Investigación en Biomedicina de Buenos Aires (IBioBA) - CONICET - Partner Institute of the Max Planck Society, Buenos Aires, Argentina.
Cardiovasc Res. 2022 Jan 7;118(1):305-315. doi: 10.1093/cvr/cvaa310.
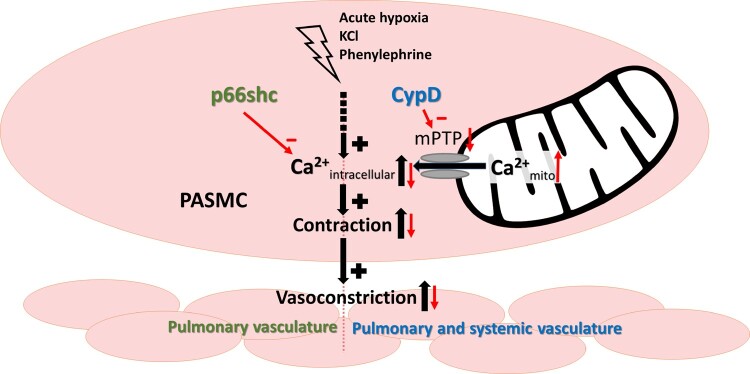
The pulmonary vascular tone and hypoxia-induced alterations of the pulmonary vasculature may be regulated by the mitochondrial membrane permeability transition pore (mPTP) that controls mitochondrial calcium load and apoptosis. We thus investigated, if the mitochondrial proteins p66shc and cyclophilin D (CypD) that regulate mPTP opening affect the pulmonary vascular tone.
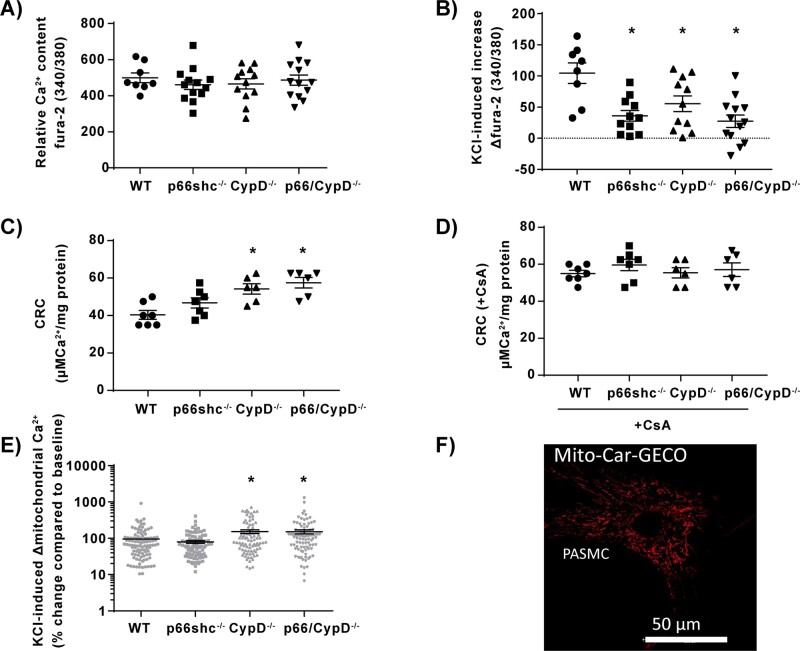
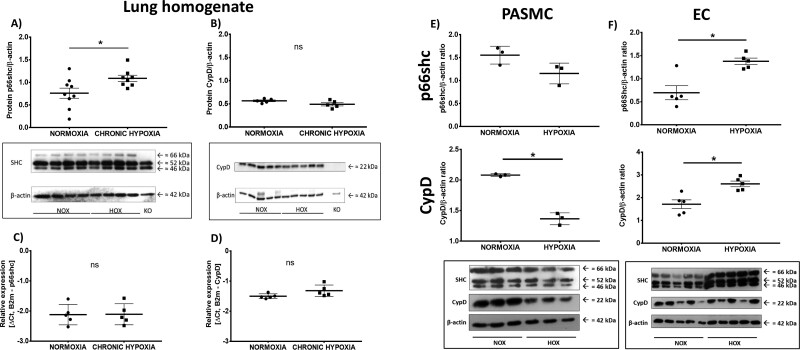
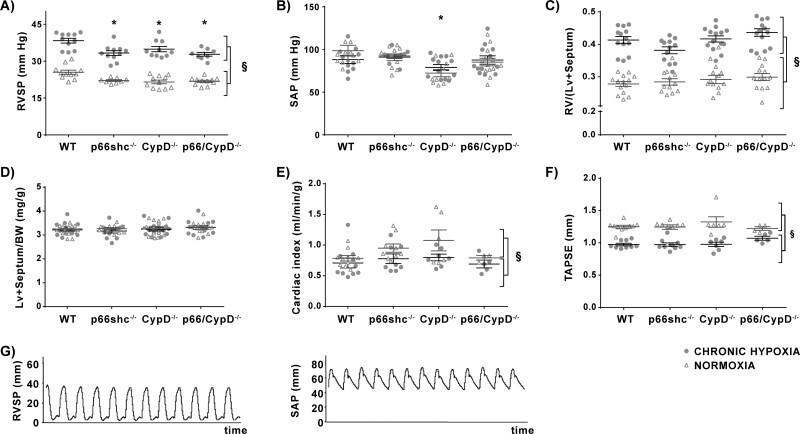
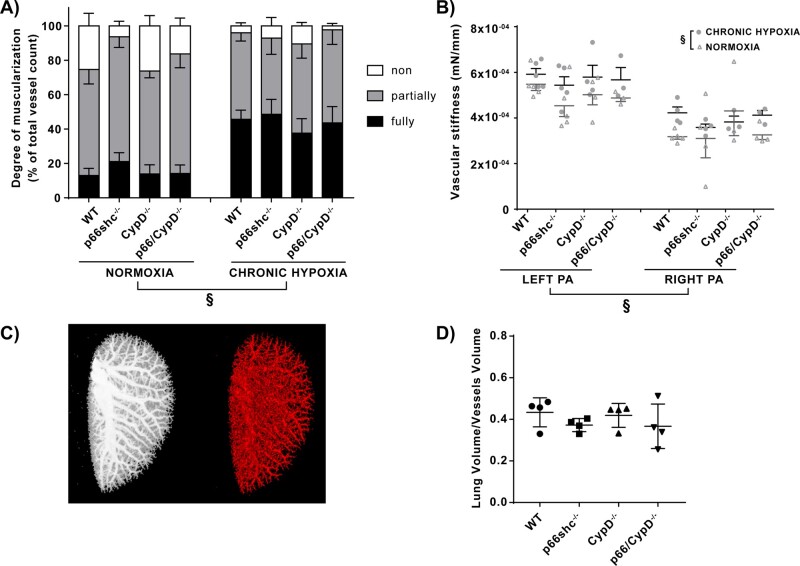
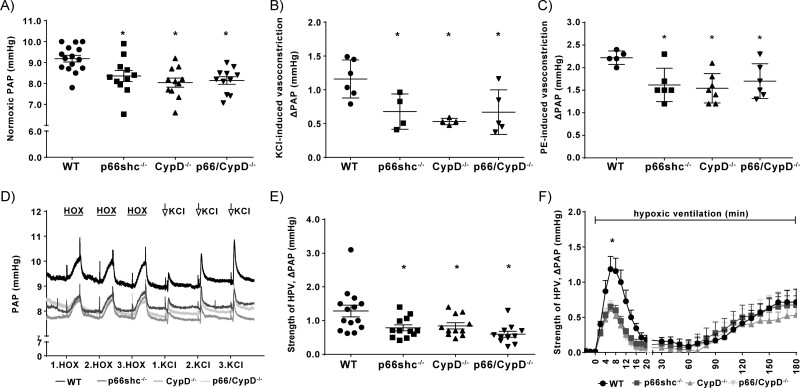
Mice deficient for p66shc (p66shc-/-), CypD (CypD-/-), or both proteins (p66shc/CypD-/-) exhibited decreased pulmonary vascular resistance (PVR) compared to wild-type mice determined in isolated lungs and in vivo. In contrast, systemic arterial pressure was only lower in CypD-/- mice. As cardiac function and pulmonary vascular remodelling did not differ between genotypes, we determined alterations of vascular contractility in isolated lungs and calcium handling in pulmonary arterial smooth muscle cells (PASMC) as underlying reason for decreased PVR. Potassium chloride (KCl)-induced pulmonary vasoconstriction and KCl-induced cytosolic calcium increase determined by Fura-2 were attenuated in all gene-deficient mice. In contrast, KCl-induced mitochondrial calcium increase determined by the genetically encoded Mito-Car-GECO and calcium retention capacity were increased only in CypD-/- and p66shc/CypD-/- mitochondria indicating that decreased mPTP opening affected KCl-induced intracellular calcium peaks in these cells. All mouse strains showed a similar pulmonary vascular response to chronic hypoxia, while acute hypoxic pulmonary vasoconstriction was decreased in gene-deficient mice indicating that CypD and p66shc regulate vascular contractility but not remodelling.
We conclude that p66shc specifically regulates the pulmonary vascular tone, while CypD also affects systemic pressure. However, only CypD acts via regulation of mPTP opening and mitochondrial calcium regulation.
肺血管张力以及低氧诱导的肺血管改变可能受线粒体膜通透性转换孔(mPTP)调控,该孔控制线粒体钙负荷和细胞凋亡。因此,我们研究了调节mPTP开放的线粒体蛋白p66shc和亲环蛋白D(CypD)是否影响肺血管张力。
与野生型小鼠相比,p66shc基因缺陷(p66shc-/-)、CypD基因缺陷(CypD-/-)或两种蛋白均缺陷(p66shc/CypD-/-)的小鼠在离体肺和体内实验中均表现出肺血管阻力(PVR)降低。相比之下,仅CypD-/-小鼠的体动脉血压较低。由于不同基因型小鼠的心脏功能和肺血管重塑无差异,我们测定了离体肺血管收缩性的改变以及肺动脉平滑肌细胞(PASMC)中钙的处理情况,以此作为PVR降低的潜在原因。在所有基因缺陷小鼠中,氯化钾(KCl)诱导的肺血管收缩以及通过Fura-2测定的KCl诱导的细胞溶质钙增加均减弱。相比之下,通过基因编码的Mito-Car-GECO测定的KCl诱导的线粒体钙增加以及钙潴留能力仅在CypD-/-和p66shc/CypD-/-线粒体中增加,这表明mPTP开放减少影响了这些细胞中KCl诱导的细胞内钙峰。所有小鼠品系对慢性低氧均表现出相似的肺血管反应,而基因缺陷小鼠的急性低氧性肺血管收缩减弱,这表明CypD和p66shc调节血管收缩性而非血管重塑。
我们得出结论,p66shc特异性调节肺血管张力,而CypD也影响体循环压力。然而,只有CypD通过调节mPTP开放和线粒体钙调节发挥作用。