Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), 13125 Berlin, Germany.
Charité Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, 10117 Berlin, Germany.
Neuron. 2021 Jan 20;109(2):299-313.e9. doi: 10.1016/j.neuron.2020.10.005. Epub 2020 Nov 5.
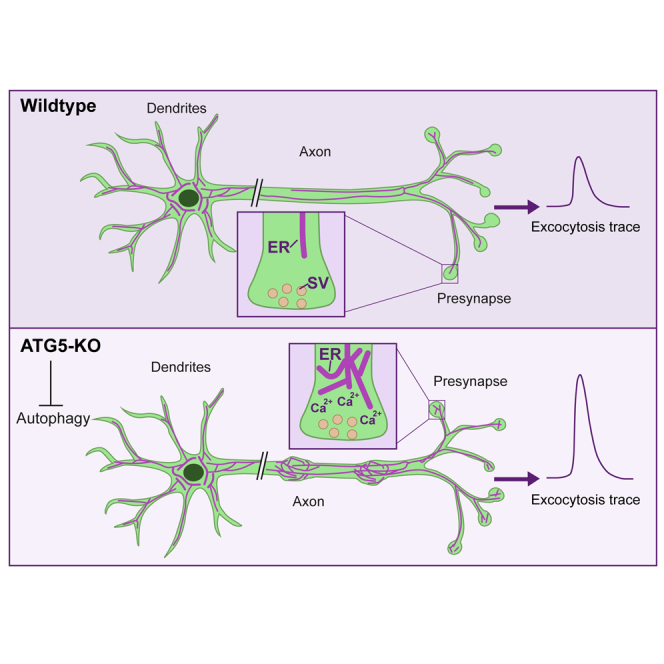
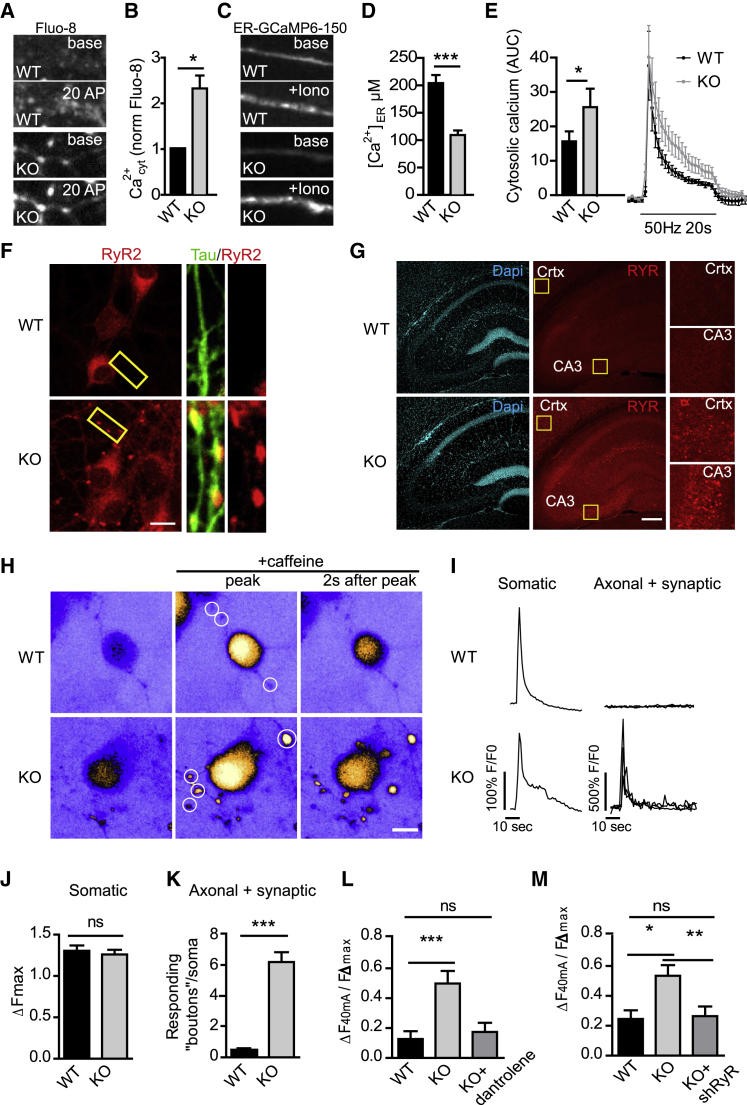
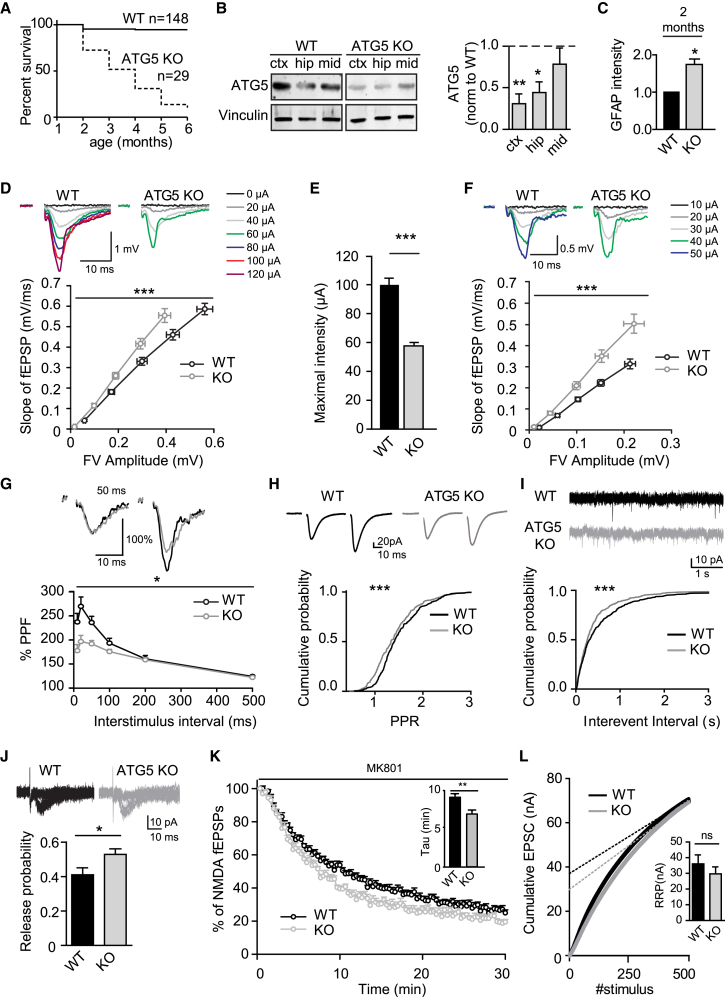
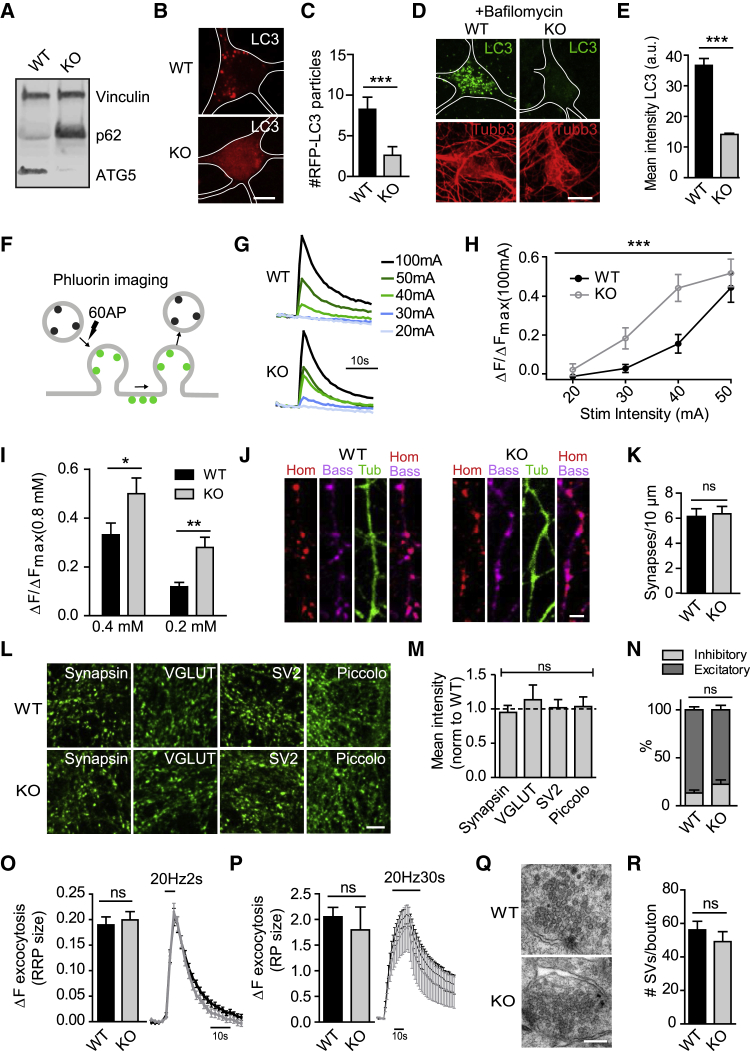
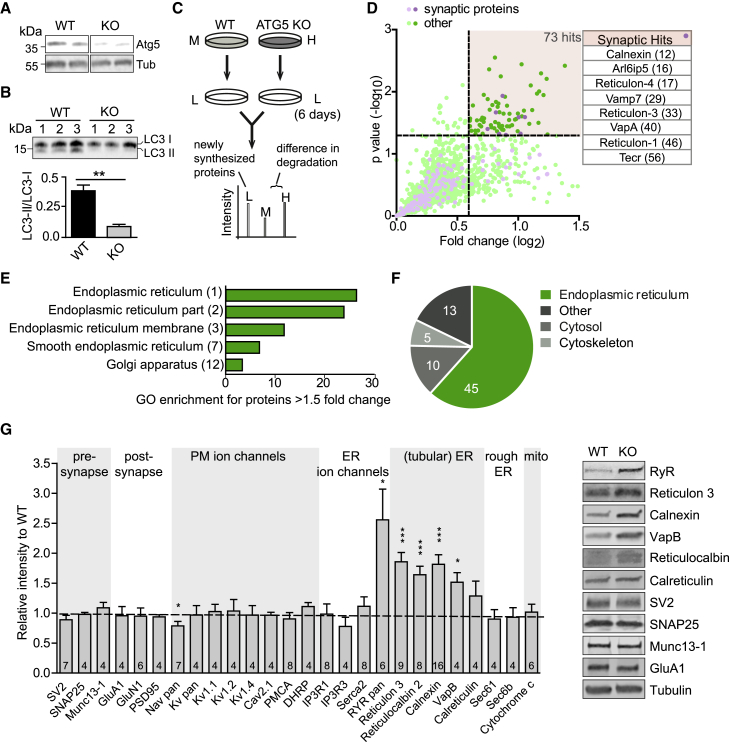
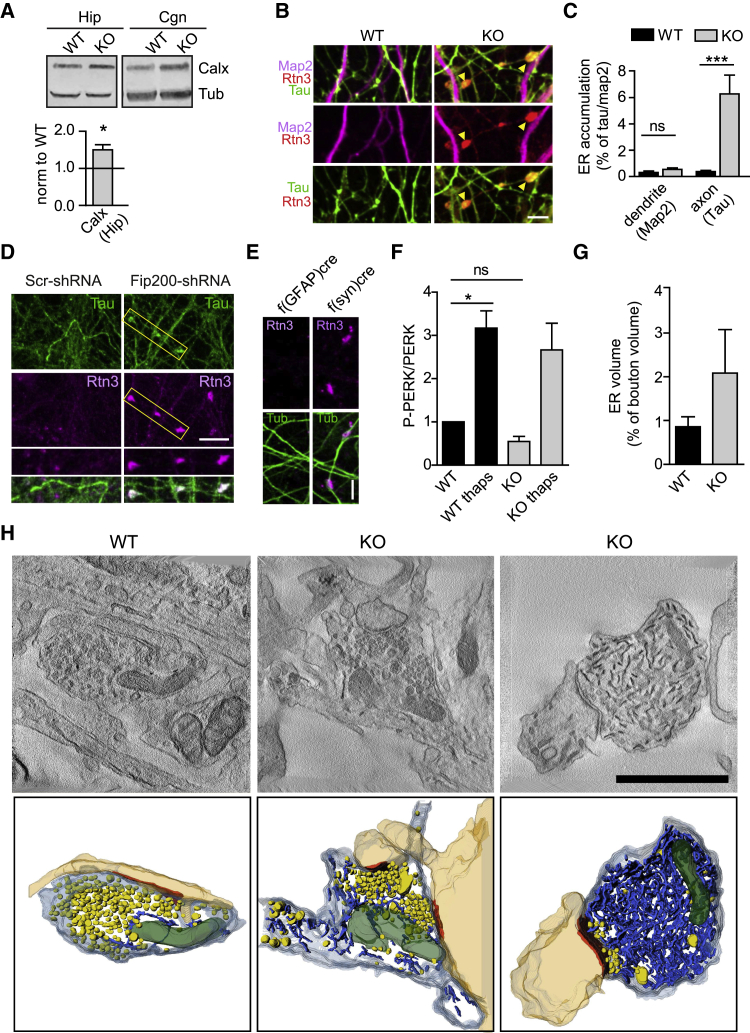
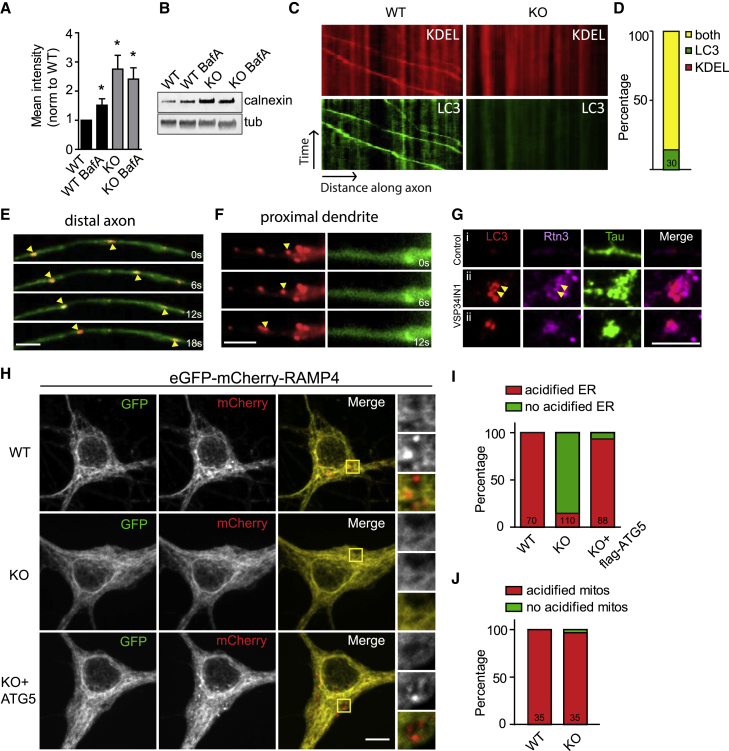
Neurons are known to rely on autophagy for removal of defective proteins or organelles to maintain synaptic neurotransmission and counteract neurodegeneration. In spite of its importance for neuronal health, the physiological substrates of neuronal autophagy in the absence of proteotoxic challenge have remained largely elusive. We use knockout mice conditionally lacking the essential autophagy protein ATG5 and quantitative proteomics to demonstrate that loss of neuronal autophagy causes selective accumulation of tubular endoplasmic reticulum (ER) in axons, resulting in increased excitatory neurotransmission and compromised postnatal viability in vivo. The gain in excitatory neurotransmission is shown to be a consequence of elevated calcium release from ER stores via ryanodine receptors accumulated in axons and at presynaptic sites. We propose a model where neuronal autophagy controls axonal ER calcium stores to regulate neurotransmission in healthy neurons and in the brain.
神经元依赖自噬作用清除缺陷蛋白或细胞器,以维持突触神经递质传递并抵抗神经退行性病变。尽管自噬作用对神经元健康很重要,但在没有蛋白毒性挑战的情况下,神经元自噬的生理底物在很大程度上仍然难以捉摸。我们使用条件性敲除必需自噬蛋白 ATG5 的小鼠和定量蛋白质组学方法证明,神经元自噬的缺失会导致轴突中管状内质网(ER)的选择性积累,从而导致兴奋性神经递质传递增加,并损害体内出生后的存活率。兴奋性神经递质传递的增加被证明是由于通过在轴突和突触前部位积累的肌醇 1,4,5-三磷酸受体(ryanodine receptors),从 ER 库中释放出更多的钙离子。我们提出了一个模型,其中神经元自噬作用控制轴突 ER 钙库,以调节健康神经元和大脑中的神经递质传递。