University of Minnesota, Minneapolis, Minnesota, United States of America.
University of Washington, Seattle, Washington, United States of America.
PLoS One. 2020 Nov 10;15(11):e0241503. doi: 10.1371/journal.pone.0241503. eCollection 2020.
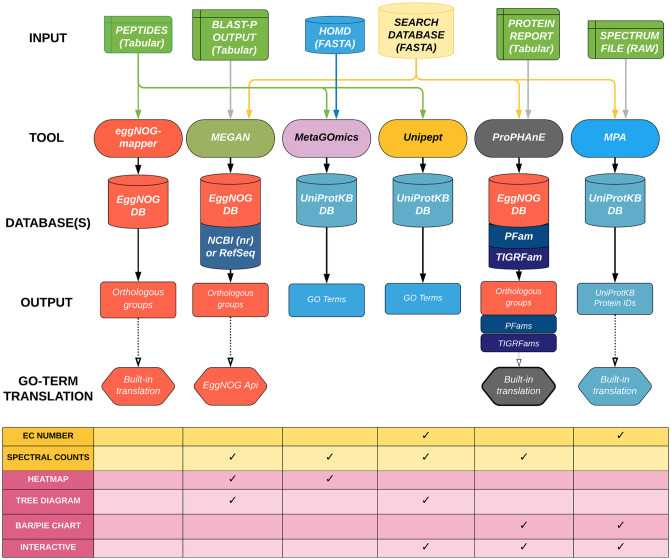
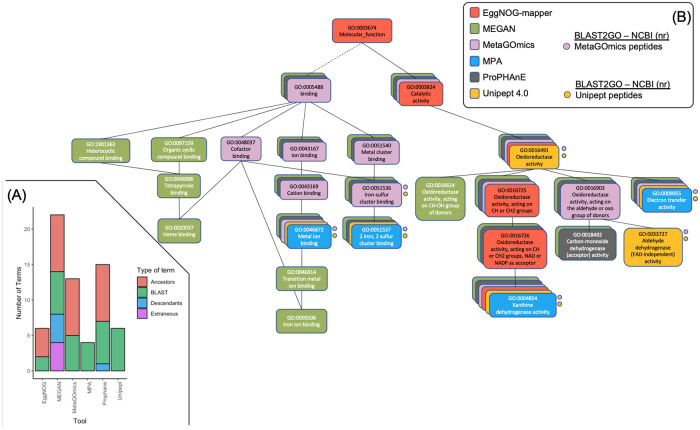
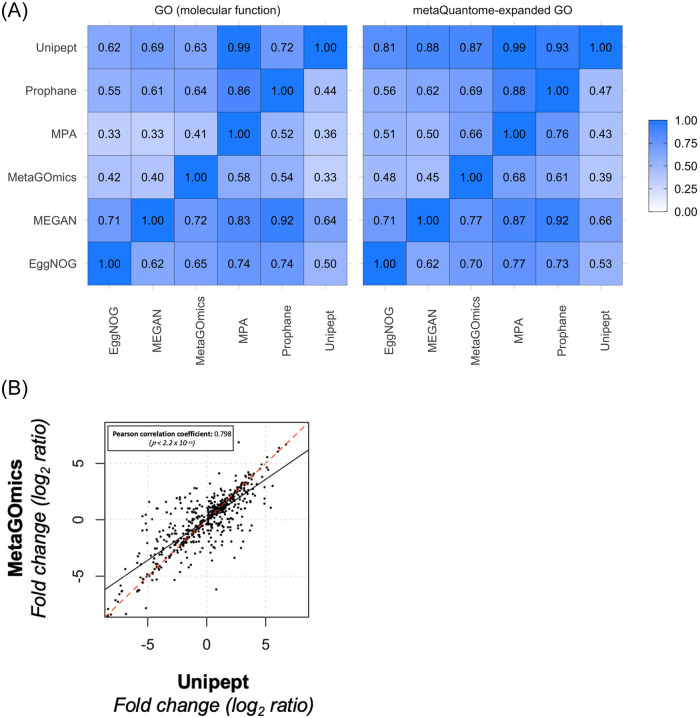
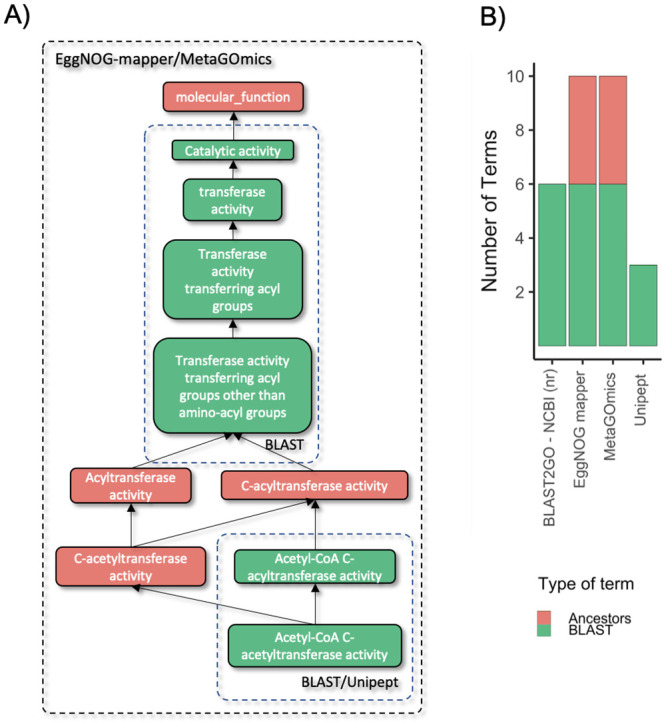
To gain a thorough appreciation of microbiome dynamics, researchers characterize the functional relevance of expressed microbial genes or proteins. This can be accomplished through metaproteomics, which characterizes the protein expression of microbiomes. Several software tools exist for analyzing microbiomes at the functional level by measuring their combined proteome-level response to environmental perturbations. In this survey, we explore the performance of six available tools, to enable researchers to make informed decisions regarding software choice based on their research goals. Tandem mass spectrometry-based proteomic data obtained from dental caries plaque samples grown with and without sucrose in paired biofilm reactors were used as representative data for this evaluation. Microbial peptides from one sample pair were identified by the X! tandem search algorithm via SearchGUI and subjected to functional analysis using software tools including eggNOG-mapper, MEGAN5, MetaGOmics, MetaProteomeAnalyzer (MPA), ProPHAnE, and Unipept to generate functional annotation through Gene Ontology (GO) terms. Among these software tools, notable differences in functional annotation were detected after comparing differentially expressed protein functional groups. Based on the generated GO terms of these tools we performed a peptide-level comparison to evaluate the quality of their functional annotations. A BLAST analysis against the NCBI non-redundant database revealed that the sensitivity and specificity of functional annotation varied between tools. For example, eggNOG-mapper mapped to the most number of GO terms, while Unipept generated more accurate GO terms. Based on our evaluation, metaproteomics researchers can choose the software according to their analytical needs and developers can use the resulting feedback to further optimize their algorithms. To make more of these tools accessible via scalable metaproteomics workflows, eggNOG-mapper and Unipept 4.0 were incorporated into the Galaxy platform.
为了深入了解微生物组动态,研究人员需要确定表达微生物基因或蛋白质的功能相关性。这可以通过宏蛋白质组学来实现,其对微生物组的蛋白质表达进行分析。目前存在几种软件工具可以通过测量微生物组对环境扰动的综合蛋白质组水平反应来对功能水平上的微生物组进行分析。在本研究中,我们探索了六种可用工具的性能,以便研究人员可以根据自己的研究目标,在软件选择方面做出明智的决策。通过将来自成对生物膜反应器中用和不用蔗糖培养的龋齿斑块样本的基于串联质谱的蛋白质组学数据用作该评估的代表性数据,来实现这一目标。使用 X! tandem search algorithm 通过 SearchGUI 鉴定一对样本中的微生物肽,并使用包括 eggNOG-mapper、MEGAN5、MetaGOmics、MetaProteomeAnalyzer (MPA)、ProPHAnE 和 Unipept 在内的软件工具对其进行功能分析,通过基因本体论 (GO) 术语生成功能注释。在这些软件工具中,在比较差异表达蛋白功能组时,检测到了功能注释的明显差异。基于这些工具生成的 GO 术语,我们进行了肽级别的比较,以评估其功能注释的质量。与 NCBI 非冗余数据库的 BLAST 分析表明,功能注释的敏感性和特异性在工具之间存在差异。例如,eggNOG-mapper 映射到最多数量的 GO 术语,而 Unipept 生成了更准确的 GO 术语。根据我们的评估,宏蛋白质组学研究人员可以根据自己的分析需求选择软件,而开发人员可以利用由此产生的反馈来进一步优化他们的算法。为了通过可扩展的宏蛋白质组学工作流程访问更多的此类工具,将 eggNOG-mapper 和 Unipept 4.0 集成到 Galaxy 平台中。