Department of Gastrointestinal Surgery, Shanghai Minimally Invasive Surgery Center, Ruijin Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai 200025, P.R. China.
Mol Med Rep. 2020 Dec;22(6):4696-4706. doi: 10.3892/mmr.2020.11568. Epub 2020 Oct 7.
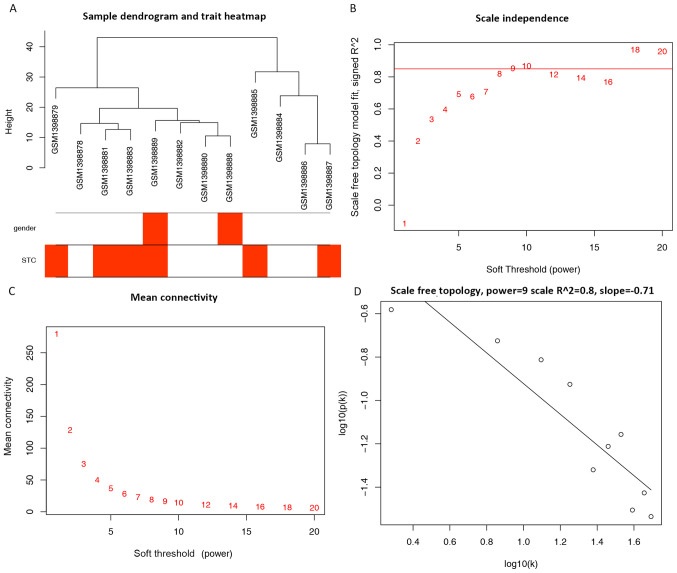
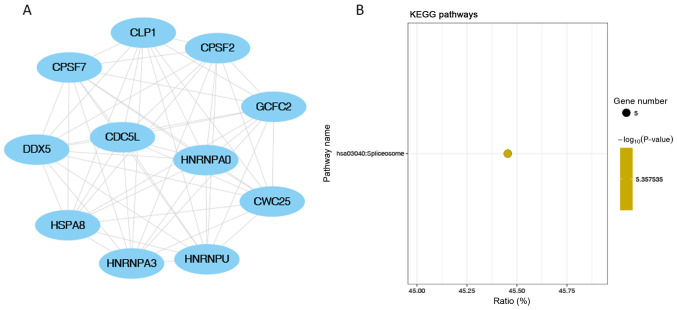
The pathogenesis of slow‑transit constipation (STC) remains largely unclear, with the roles of microRNAs (miRs/miRNAs) yet to be determined. Co‑expression network analysis of miRNAs in STC is crucial to elucidating potential underlying mechanisms. Weighted gene correlation network analysis was performed in the miRNA expression profile of STC (GSE57969). The key miRNA target genes were further functionally enriched by Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO). A Protein‑Protein Interaction (PPI) network was constructed, with a total of 12 color‑clustered modules determined. Seven key miRNAs were established, including five miRNAs from the turquoise module (hsa‑miR‑20b, hsa‑miR‑128, hsa‑miR‑129‑3p, hsa‑miR‑30b and hsa‑miR‑340), one miRNA from the blue module (hsa‑miR‑619) and one from the black module (hsa‑miR‑486‑3p). A total of 2,077 key miRNA target genes were predicted. GO analysis revealed that the 'protein modification process' and 'cellular protein modification process' were the most significantly enriched processes in the 'Biological Processes' category, whereas the 'nucleoplasm' in 'Cellular Components' and 'enzyme binding' in 'Molecular Functions' were the most significantly enriched processes. The 'cAMP signalling pathway' was the top KEGG pathway. The hub genes identified from the PPI network included calmodulin (CALM)2, CALM1, histone deacetylase (HDAC)3, glycogen synthase kinase 3 β, HDAC9, heat‑shock protein family A member 8, G‑protein subunit γ (GNG)13, HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1, GNG10 and GNG7. This bioinformatics analysis demonstrated co‑expressed miRNA networks with insightful genes and pathways associated with STC.
慢传输型便秘(STC)的发病机制尚不清楚,miRNAs(miRs/miRNAs)的作用尚待确定。对 STC 中 miRNA 的共表达网络分析对于阐明潜在的机制至关重要。对 STC 的 miRNA 表达谱(GSE57969)进行了加权基因相关网络分析。进一步通过京都基因与基因组百科全书(KEGG)和基因本体论(GO)对关键 miRNA 靶基因进行功能富集。构建了一个蛋白质-蛋白质相互作用(PPI)网络,共确定了 12 个颜色聚类模块。确定了 7 个关键 miRNA,包括来自绿松石模块的 5 个 miRNA(hsa-miR-20b、hsa-miR-128、hsa-miR-129-3p、hsa-miR-30b 和 hsa-miR-340)、来自蓝色模块的 1 个 miRNA(hsa-miR-619)和来自黑色模块的 1 个 miRNA(hsa-miR-486-3p)。共预测了 2077 个关键 miRNA 靶基因。GO 分析显示,“蛋白质修饰过程”和“细胞蛋白质修饰过程”是“生物过程”类别中最显著富集的过程,而“核质”在“细胞成分”和“酶结合”在“分子功能”中是最显著富集的过程。KEGG 途径中排名最高的是“cAMP 信号通路”。PPI 网络中鉴定的枢纽基因包括钙调蛋白(CALM)2、CALM1、组蛋白去乙酰化酶(HDAC)3、糖原合酶激酶 3β、HDAC9、热休克蛋白家族 A 成员 8、G-蛋白亚基γ(GNG)13、HECT 结构域和含锚蛋白重复的 E3 泛素蛋白连接酶 1、GNG10 和 GNG7。这项生物信息学分析表明,与 STC 相关的 miRNA 网络存在共表达基因和途径。