Genetics Unit, Universitat Pompeu Fabra, Barcelona 08003, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Barcelona 08003, Spain.
Genome Res. 2020 Dec;30(12):1802-1814. doi: 10.1101/gr.258301.119. Epub 2020 Nov 17.
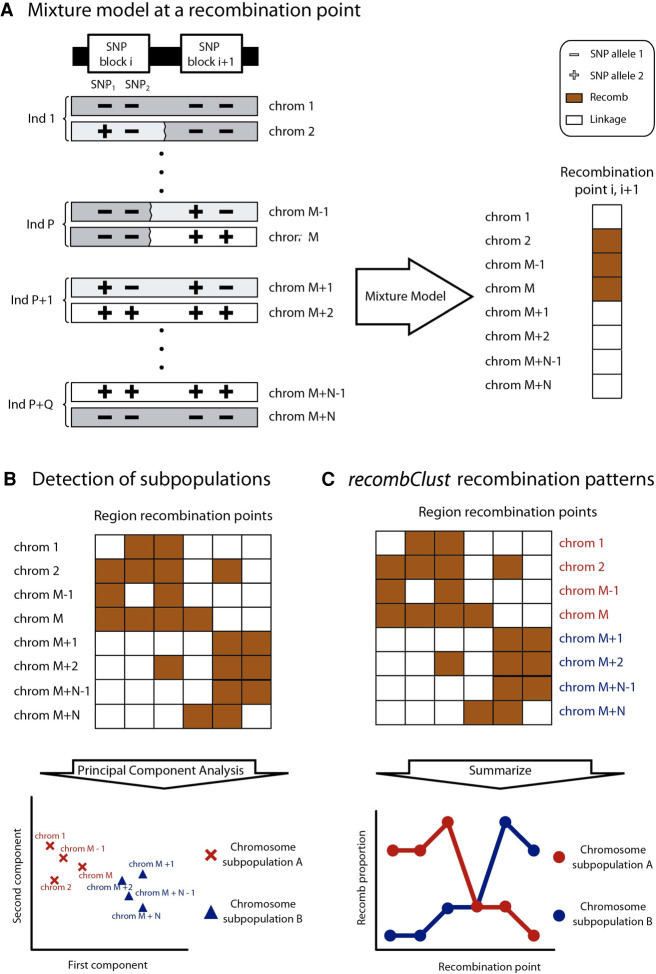
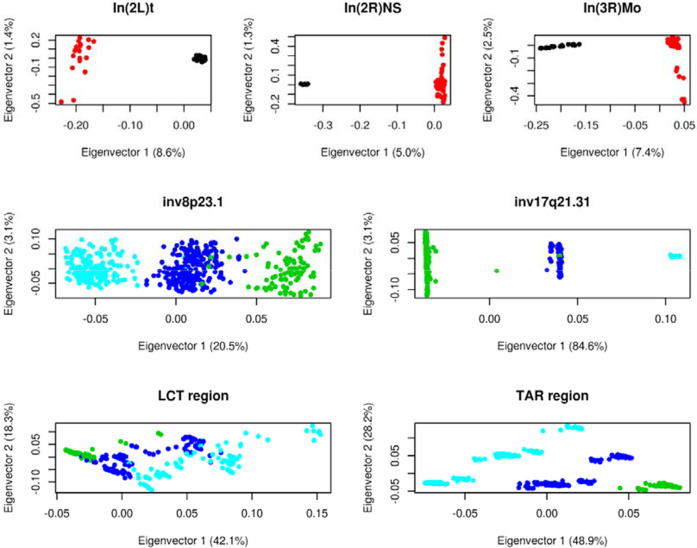
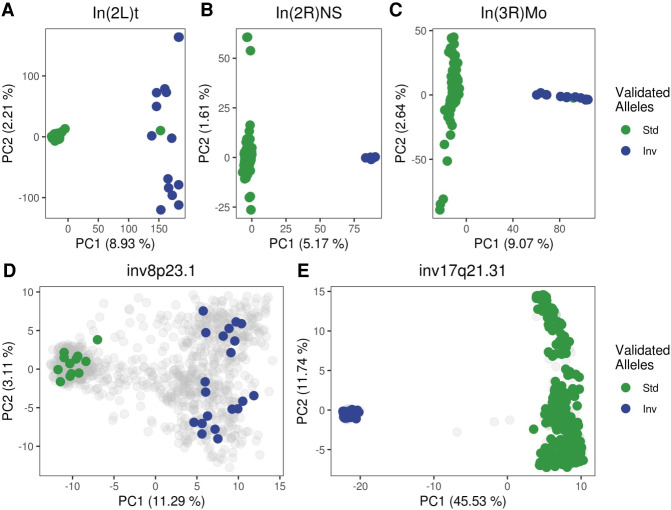
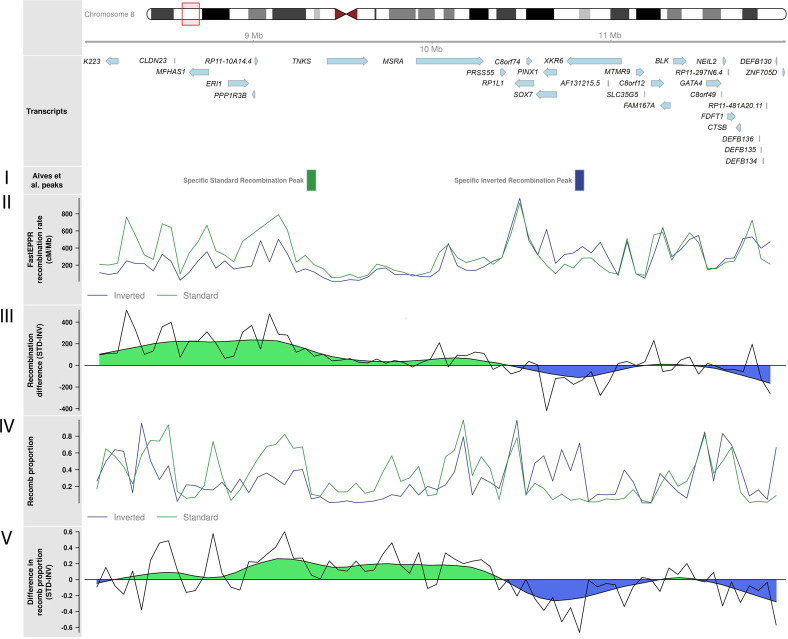
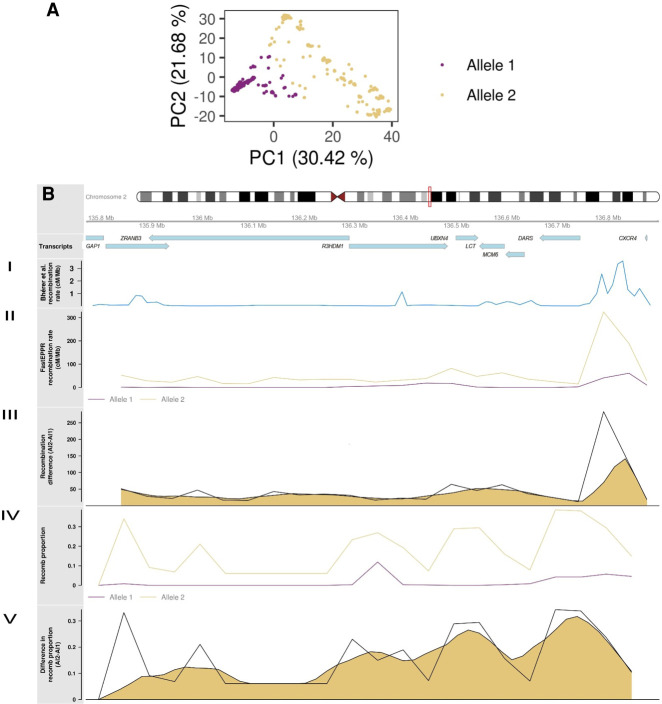
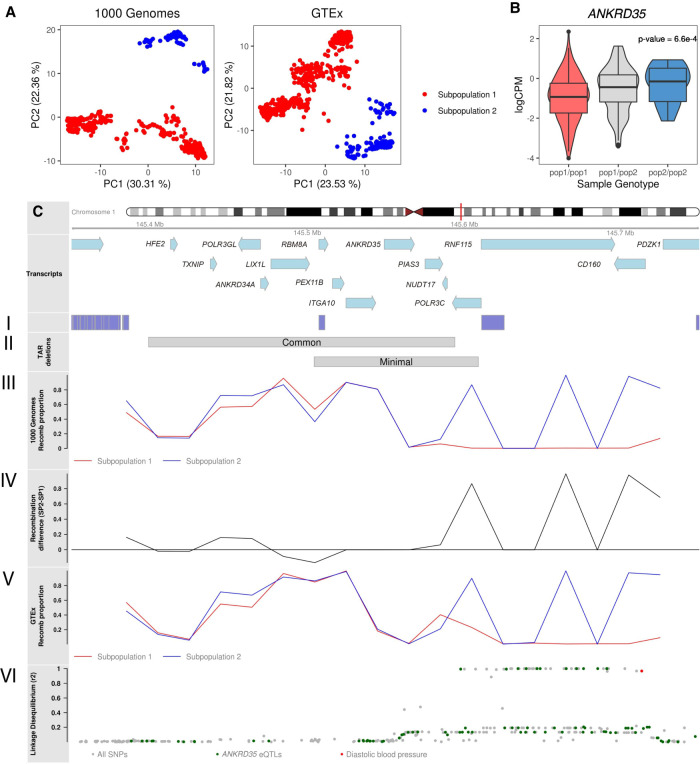
Recombination is a main source of genetic variability. However, the potential role of the variation generated by recombination in phenotypic traits, including diseases, remains unexplored because there is currently no method to infer chromosomal subpopulations based on recombination pattern differences. We developed , a method that uses SNP-phased data to detect differences in historic recombination in a chromosome population. We validated our method by performing simulations and by using real data to accurately predict the alleles of well-known recombination modifiers, including common inversions in and human, and the chromosomes under selective pressure at the lactase locus in humans. We then applied to the complex human 1q21.1 region, where nonallelic homologous recombination produces deleterious phenotypes. We discovered and validated the presence of two different recombination histories in these regions that significantly associated with the differential expression of in whole blood and that were in high linkage with variants previously associated with hypertension. By detecting differences in historic recombination, our method opens a way to assess the influence of recombination variation in phenotypic traits.
重组是遗传变异的主要来源。然而,由于目前尚无基于重组模式差异推断染色体亚群的方法,因此重组产生的变异在表型特征(包括疾病)中的潜在作用仍未得到探索。我们开发了一种方法,该方法使用 SNP 相位数据来检测染色体群体中历史重组的差异。我们通过执行模拟并使用真实数据来准确预测已知重组修饰因子的等位基因来验证我们的方法,这些因子包括 和人类中的常见倒位,以及人类乳糖酶基因座中受选择压力影响的染色体。然后,我们将 应用于复杂的人类 1q21.1 区域,在该区域中非等位基因同源重组会产生有害的表型。我们发现并验证了这些区域中两种不同的重组历史的存在,它们与全血中 的差异表达显著相关,并且与先前与高血压相关的变体高度连锁。通过检测历史重组的差异,我们的方法为评估重组变异对表型特征的影响开辟了道路。