Słabicki Mikołaj, Yoon Hojong, Koeppel Jonas, Nitsch Lena, Roy Burman Shourya S, Di Genua Cristina, Donovan Katherine A, Sperling Adam S, Hunkeler Moritz, Tsai Jonathan M, Sharma Rohan, Guirguis Andrew, Zou Charles, Chudasama Priya, Gasser Jessica A, Miller Peter G, Scholl Claudia, Fröhling Stefan, Nowak Radosław P, Fischer Eric S, Ebert Benjamin L
Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Nature. 2020 Dec;588(7836):164-168. doi: 10.1038/s41586-020-2925-1. Epub 2020 Nov 18.
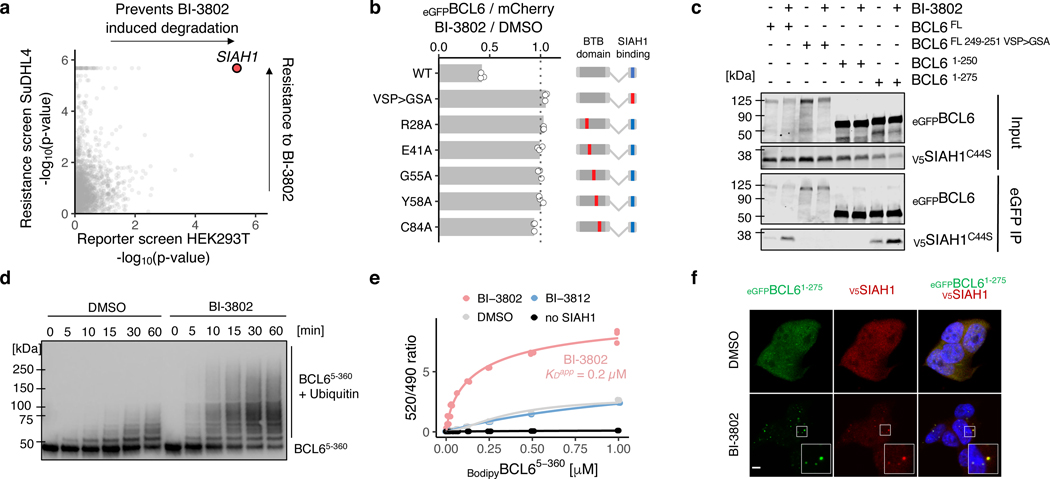
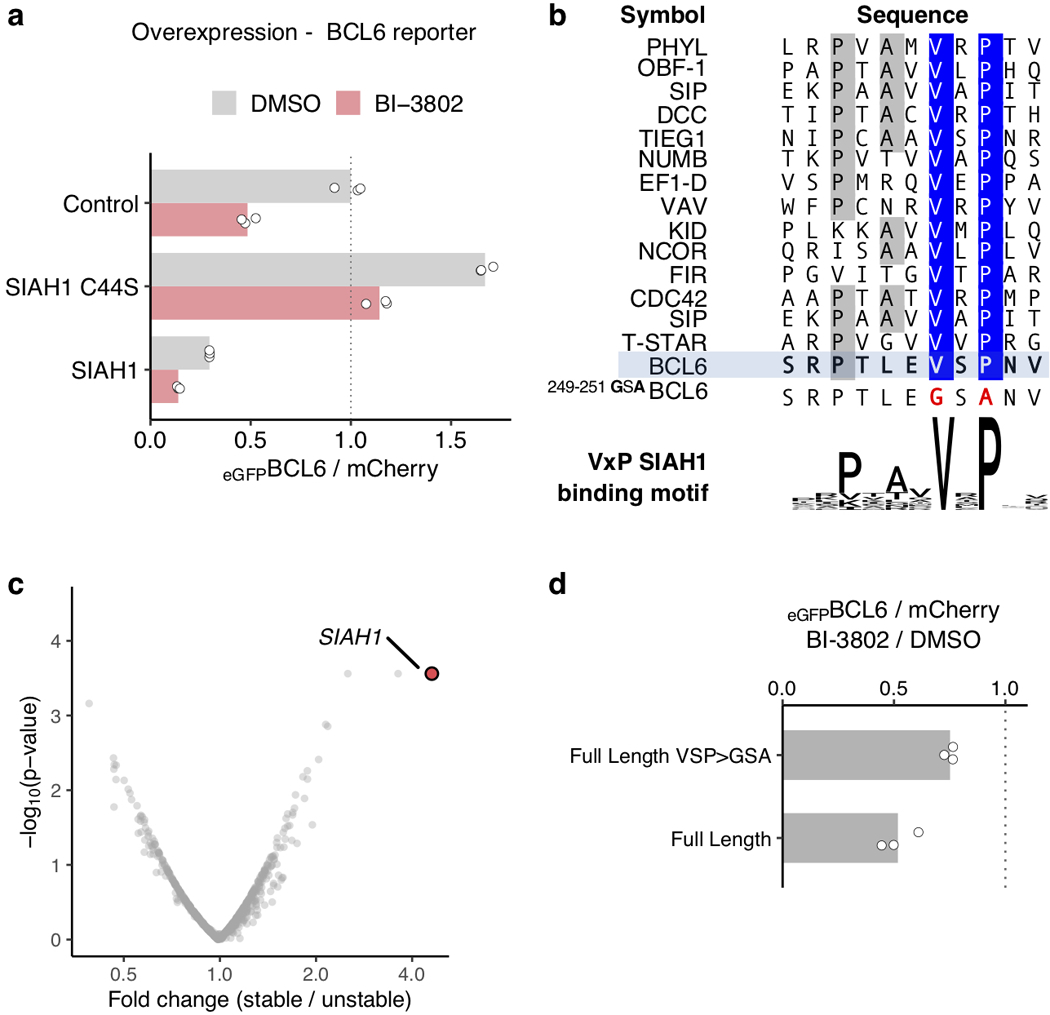
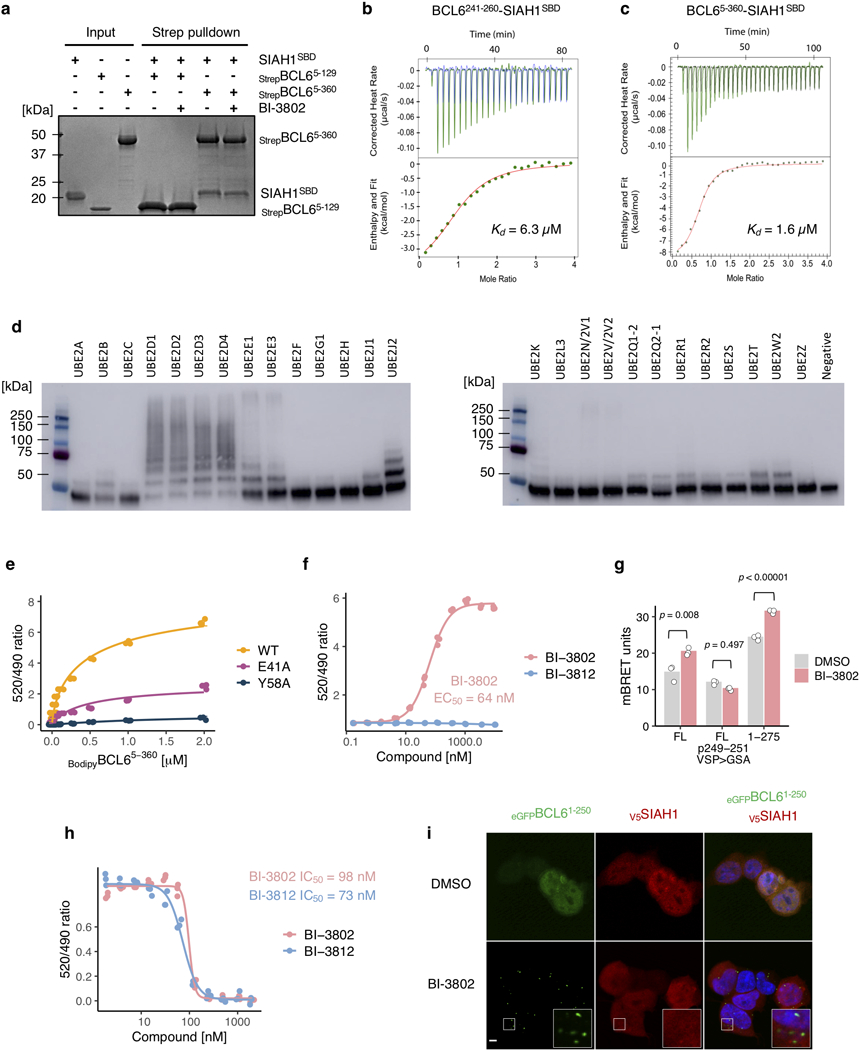
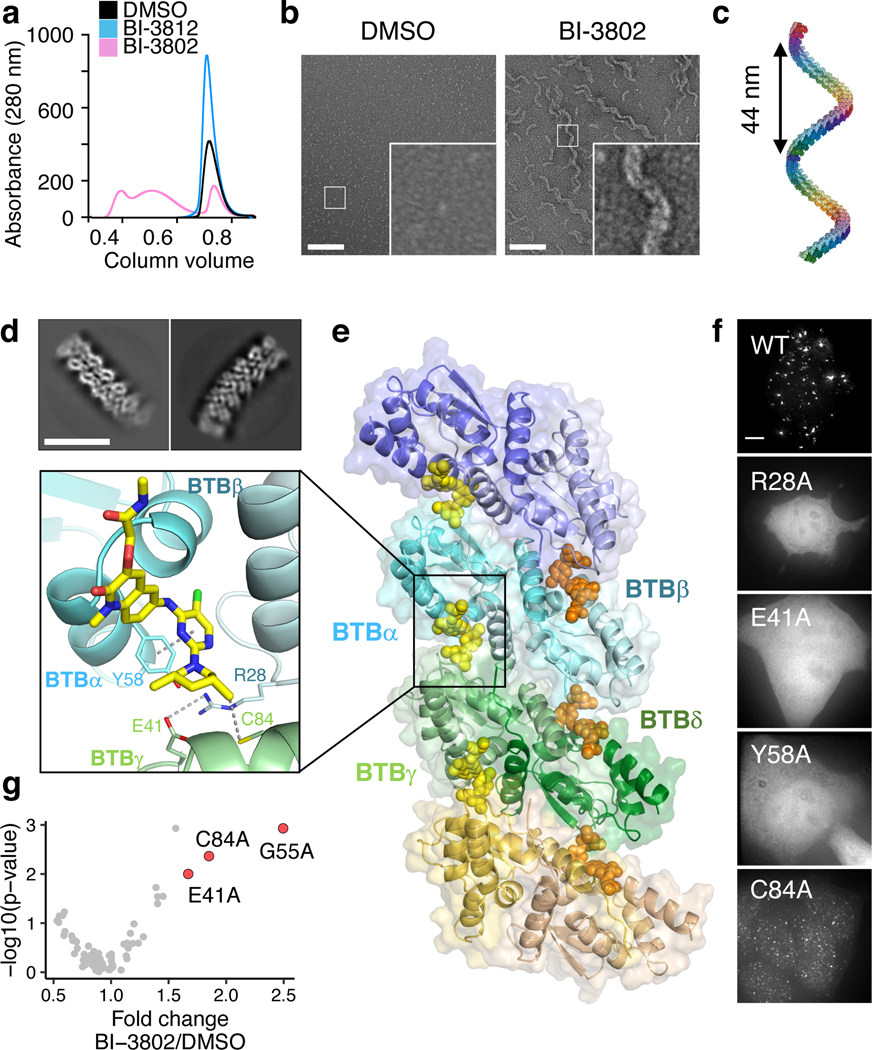
Effective and sustained inhibition of non-enzymatic oncogenic driver proteins is a major pharmacological challenge. The clinical success of thalidomide analogues demonstrates the therapeutic efficacy of drug-induced degradation of transcription factors and other cancer targets, but a substantial subset of proteins are resistant to targeted degradation using existing approaches. Here we report an alternative mechanism of targeted protein degradation, in which a small molecule induces the highly specific, reversible polymerization of a target protein, followed by its sequestration into cellular foci and subsequent degradation. BI-3802 is a small molecule that binds to the Broad-complex, Tramtrack and Bric-à-brac (BTB) domain of the oncogenic transcription factor B cell lymphoma 6 (BCL6) and leads to the proteasomal degradation of BCL6. We use cryo-electron microscopy to reveal how the solvent-exposed moiety of a BCL6-binding molecule contributes to a composite ligand-protein surface that engages BCL6 homodimers to form a supramolecular structure. Drug-induced formation of BCL6 filaments facilitates ubiquitination by the SIAH1 E3 ubiquitin ligase. Our findings demonstrate that a small molecule such as BI-3802 can induce polymerization coupled to highly specific protein degradation, which in the case of BCL6 leads to increased pharmacological activity compared to the effects induced by other BCL6 inhibitors. These findings open new avenues for the development of therapeutic agents and synthetic biology.
有效且持续地抑制非酶促致癌驱动蛋白是一项重大的药理学挑战。沙利度胺类似物的临床成功证明了药物诱导转录因子和其他癌症靶点降解的治疗效果,但相当一部分蛋白质对现有方法的靶向降解具有抗性。在此,我们报告了一种靶向蛋白降解的替代机制,即小分子诱导靶蛋白高度特异性、可逆的聚合,随后将其隔离到细胞灶中并进行后续降解。BI-3802是一种小分子,它与致癌转录因子B细胞淋巴瘤6(BCL6)的广泛复合体、Tramtrack和Bric-à-brac(BTB)结构域结合,并导致BCL6的蛋白酶体降解。我们使用冷冻电子显微镜揭示了BCL6结合分子的溶剂暴露部分如何促成一个复合配体-蛋白表面,该表面与BCL6同二聚体结合形成超分子结构。药物诱导的BCL6细丝形成促进了SIAH1 E3泛素连接酶介导的泛素化。我们的研究结果表明,像BI-3802这样的小分子可以诱导聚合并伴随高度特异性的蛋白降解,就BCL6而言,与其他BCL6抑制剂诱导的效应相比,这会导致药理活性增加。这些发现为治疗药物和合成生物学的发展开辟了新途径。