Department of Pathogen Biology, Medical College, Nantong University, Nantong, Jiangsu, China.
Department of Traditional Chinese Medicine, Affiliated Hospital of Nantong University, Nantong, Jiangsu, China.
Cell Death Dis. 2020 Nov 27;11(11):1016. doi: 10.1038/s41419-020-03222-1.
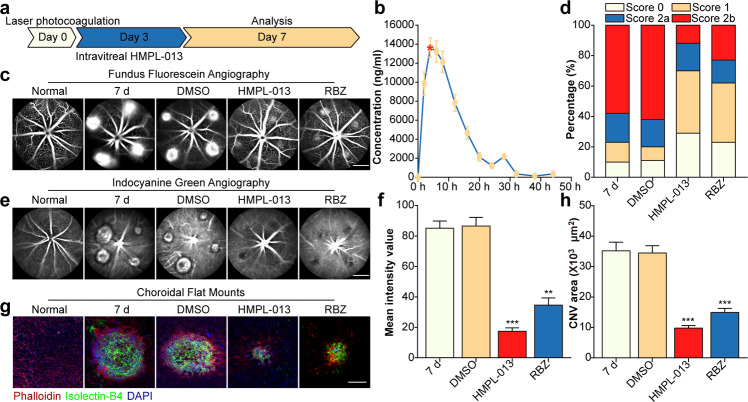
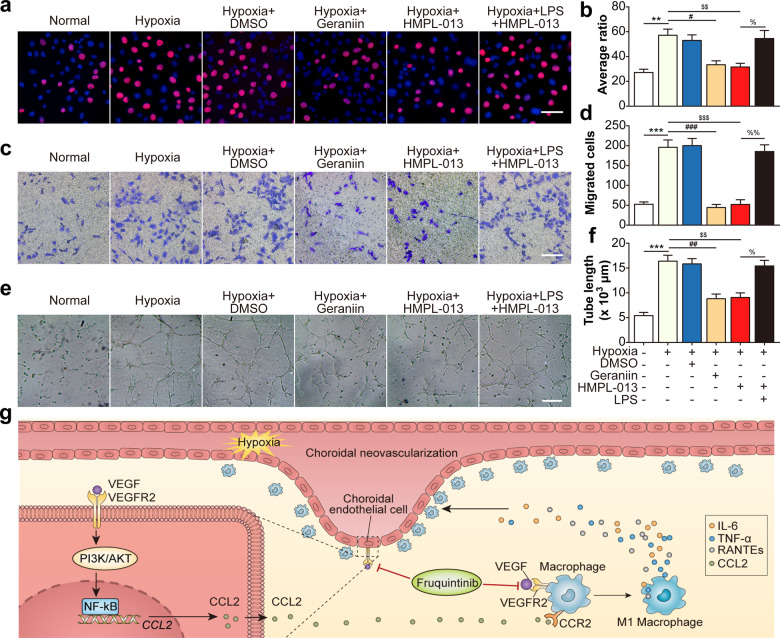
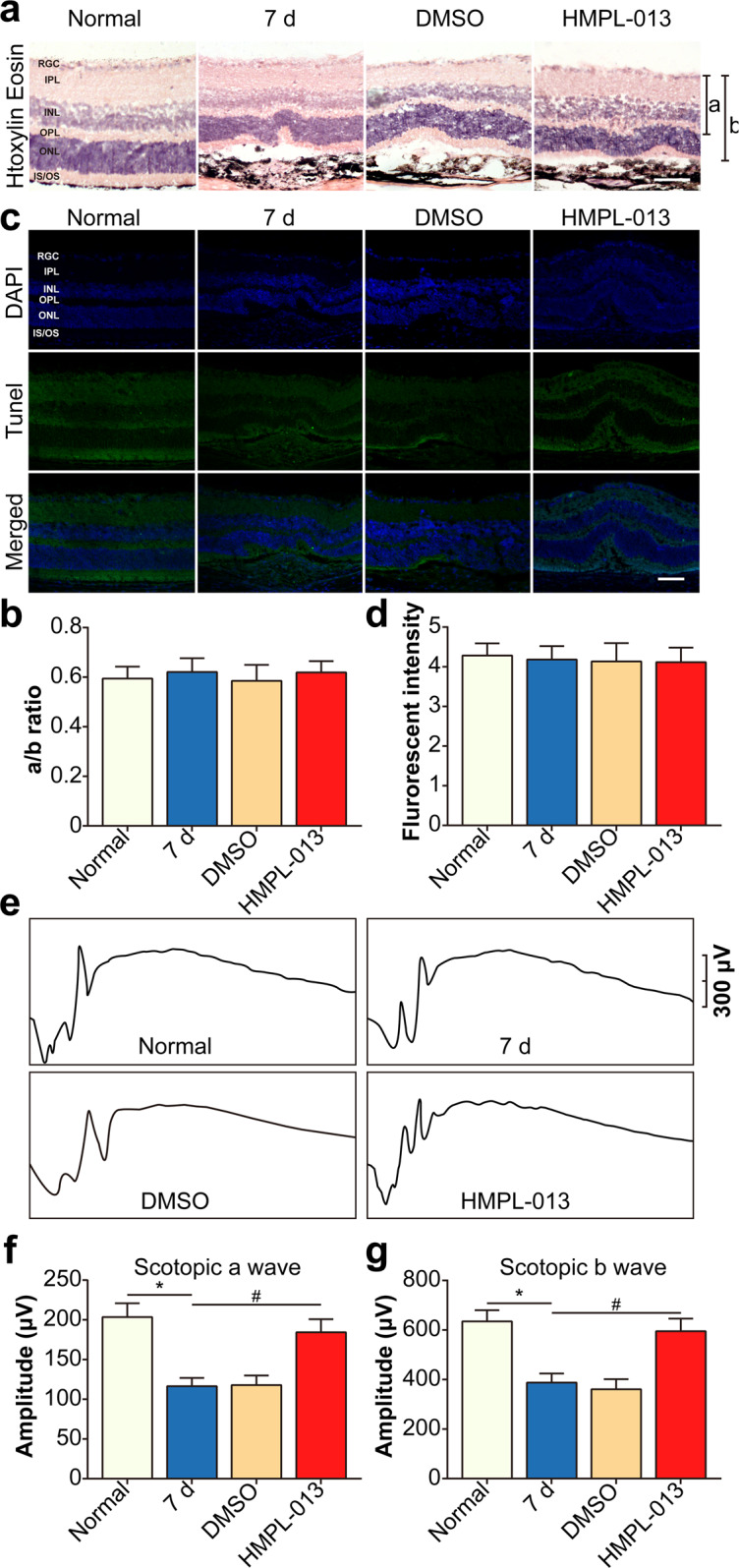
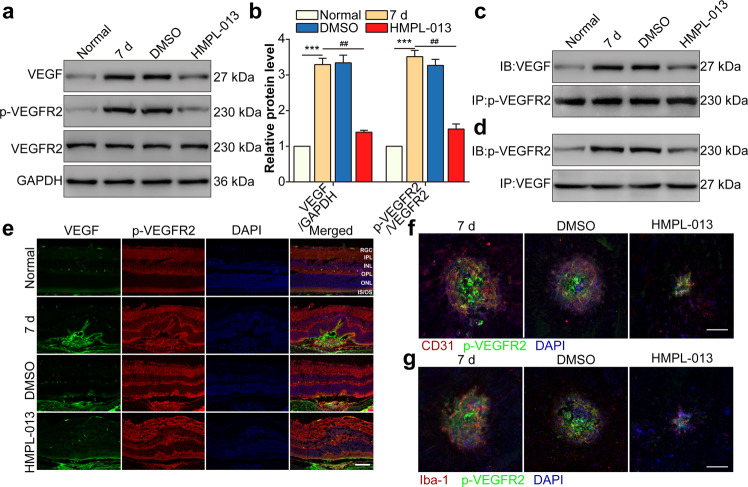
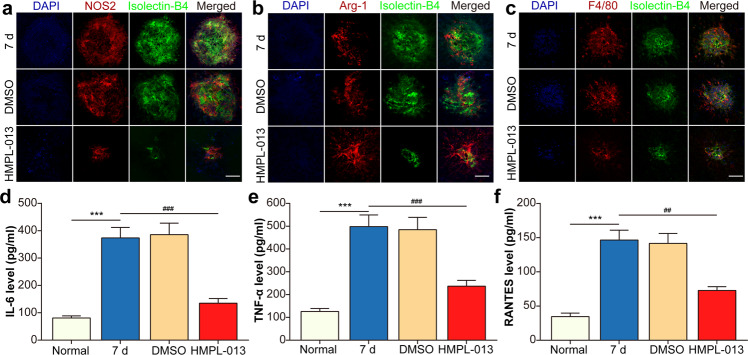
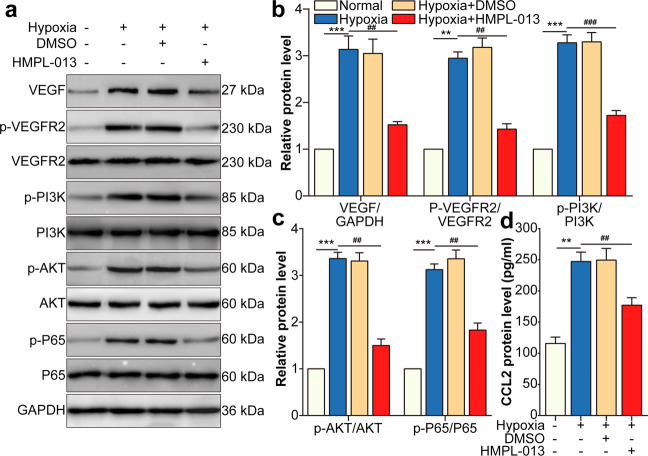
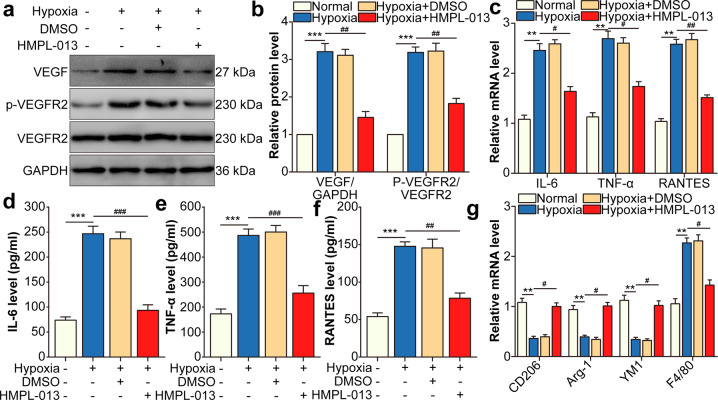
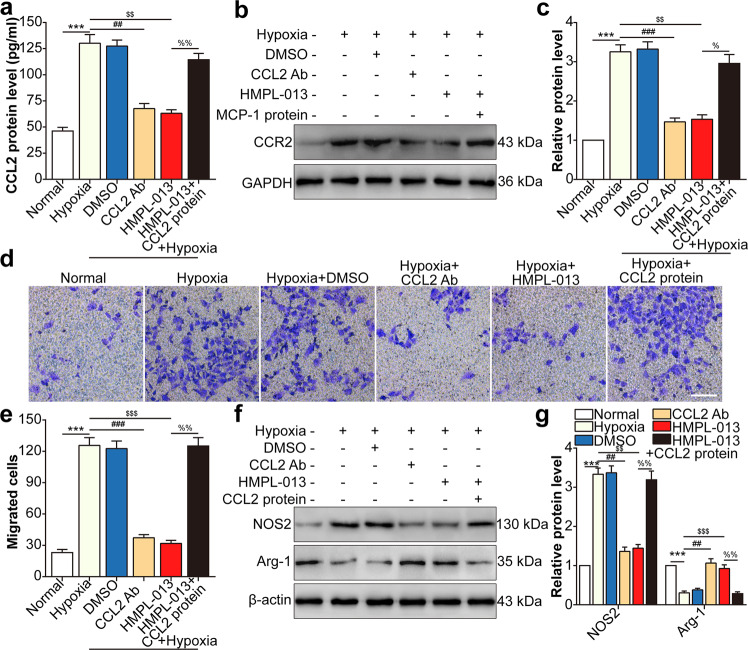
Wet age-related macular degeneration, which is characterized by choroidal neovascularization (CNV) and induces obvious vision loss. Vascular endothelial growth factor (VEGF) family member VEGF-A (also named as VEGF) and its receptor VEGFR2 contribute to the pathogenesis of CNV. Choroidal endothelial cells (CECs) secret C-C motif chemokine ligand 2 (CCL2), which attracts macrophages to CNV lesion and promotes macrophage M1 polarization. Accordingly, infiltrating macrophages secret inflammatory cytokines to promote CNV. In vivo, intravitreal injection of fruquintinib (HMPL-013), an antitumor neovascularization drug, alleviated mouse CNV formation without obvious ocular toxicity. Meanwhile, HMPL-013 inhibited VEGF/VEGFR2 binding in CECs and macrophages, as well as macrophage M1 polarization. In vitro, noncontact coculture of human choroidal vascular endothelial cells (HCVECs) and macrophages under hypoxia conditions was established. HMPL-013 downregulated VEGF/VEGFR2/phosphoinositide-3-kinase/protein kinase B (AKT)/nuclear factor kappa B pathway and CCL2 secretion in HCVECs, as well as VEGF/VEGFR2-induced macrophage M1 polarization under hypoxia condition. In addition, HMPL-013 inhibited HCEVC derived CCL2-induced macrophage migration and M1 polarization, along with macrophage M1 polarization-induced HCVECs proliferation, migration, and tube formation. Altogether, HMPL-013 alleviated CNV formation might via breaking detrimental cross talk between CECs and macrophages.
湿性年龄相关性黄斑变性,其特征为脉络膜新生血管(CNV),并导致明显的视力丧失。血管内皮生长因子(VEGF)家族成员 VEGF-A(也称为 VEGF)及其受体 VEGFR2 参与 CNV 的发病机制。脉络膜内皮细胞(CECs)分泌 C-C 基序趋化因子配体 2(CCL2),吸引巨噬细胞进入 CNV 病变并促进巨噬细胞 M1 极化。因此,浸润的巨噬细胞分泌炎症细胞因子以促进 CNV。在体内,抗肿瘤血管生成药物 fruquintinib(HMPL-013)玻璃体腔内注射可减轻小鼠 CNV 的形成,而无明显的眼毒性。同时,HMPL-013 抑制了 CECs 和巨噬细胞中的 VEGF/VEGFR2 结合以及巨噬细胞 M1 极化。在体外,建立了人脉络膜血管内皮细胞(HCVECs)和巨噬细胞在缺氧条件下的非接触共培养模型。HMPL-013 下调了 HCVECs 中的 VEGF/VEGFR2/磷酸肌醇 3-激酶/蛋白激酶 B(AKT)/核因子 kappa B 通路和 CCL2 的分泌,以及缺氧条件下 VEGF/VEGFR2 诱导的巨噬细胞 M1 极化。此外,HMPL-013 抑制了 HCEVC 衍生的 CCL2 诱导的巨噬细胞迁移和 M1 极化,以及巨噬细胞 M1 极化诱导的 HCVECs 增殖、迁移和管形成。总之,HMPL-013 通过阻断 CECs 和巨噬细胞之间的有害串扰来减轻 CNV 的形成。