Department of Pulmonary and Critical Care Medicine, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul, 05505, Republic of Korea.
Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea.
Sci Rep. 2020 Dec 3;10(1):21137. doi: 10.1038/s41598-020-78140-5.
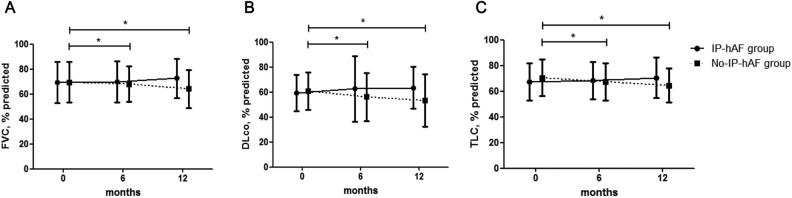
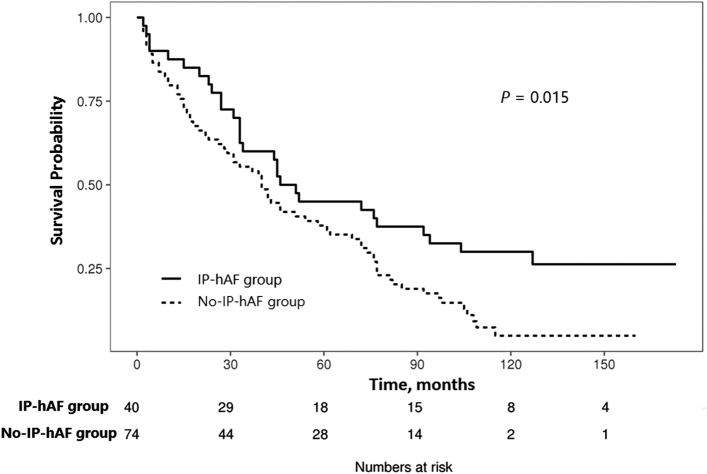
Some patients with idiopathic pulmonary fibrosis (IPF) have histopathologic features suggesting connective tissue disease (CTD); however, their clinical course and prognosis remain unclear. Thus, we aimed to investigate the clinical course and prognosis of these patients with histologic autoimmune features. Among 114 patients with biopsy-proven IPF, the histologic features were semi-quantitatively graded, and CTD scores (range: 0-9) were calculated as the sum of each score of plasma cell infiltration, lymphoid aggregates, and germinal centres. Patients with high CTD scores (≥ 4) were classified into the interstitial pneumonia with histologic autoimmune features (IP-hAF) group. The mean age of the patients was 60.0 years; 74.6% were men, 69.3% were ever-smokers, and 35.1% had IP-hAF. During follow-up, the IP-hAF group showed slower decline in lung function, and better prognosis (median survival, 48.7 vs. 40.4 months; p = 0.015) than the no-IP-hAF group. On multivariate Cox analysis, IP-hAF was an independent prognostic factor (hazard ratio, 0.522; p = 0.016), along with the lower diffusing capacity for carbon monoxide, higher scores of reticulation and honeycombing, and usual interstitial pneumonia pattern on high-resolution computed tomography. Patients with IPF having histologic autoimmune features show distinct clinical characteristics and better outcome than those without histologic autoimmune features.
一些特发性肺纤维化(IPF)患者具有提示结缔组织疾病(CTD)的组织病理学特征;然而,他们的临床过程和预后仍不清楚。因此,我们旨在研究具有组织学自身免疫特征的这些患者的临床过程和预后。在 114 例经活检证实的 IPF 患者中,对组织学特征进行半定量分级,并计算 CTD 评分(范围:0-9),即浆细胞浸润、淋巴聚集和生发中心的每个评分之和。高 CTD 评分(≥4)的患者被归类为具有组织学自身免疫特征的间质性肺炎(IP-hAF)组。患者的平均年龄为 60.0 岁;74.6%为男性,69.3%为曾吸烟者,35.1%为 IP-hAF。在随访期间,IP-hAF 组的肺功能下降速度较慢,预后较好(中位生存时间,48.7 与 40.4 个月;p=0.015)。多变量 Cox 分析显示,IP-hAF 是独立的预后因素(风险比,0.522;p=0.016),与一氧化碳弥散量降低、网状影和蜂窝影评分较高以及高分辨率计算机断层扫描上的普通间质性肺炎模式有关。具有组织学自身免疫特征的 IPF 患者表现出明显的临床特征和较好的预后,与不具有组织学自身免疫特征的患者相比。