Lananna Brian V, McKee Celia A, King Melvin W, Del-Aguila Jorge L, Dimitry Julie M, Farias Fabiana H G, Nadarajah Collin J, Xiong David D, Guo Chun, Cammack Alexander J, Elias Jack A, Zhang Jinsong, Cruchaga Carlos, Musiek Erik S
Department of Neurology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Department of Psychiatry, Washington University School of Medicine, St. Louis, MO 63110, USA.
Sci Transl Med. 2020 Dec 16;12(574). doi: 10.1126/scitranslmed.aax3519.
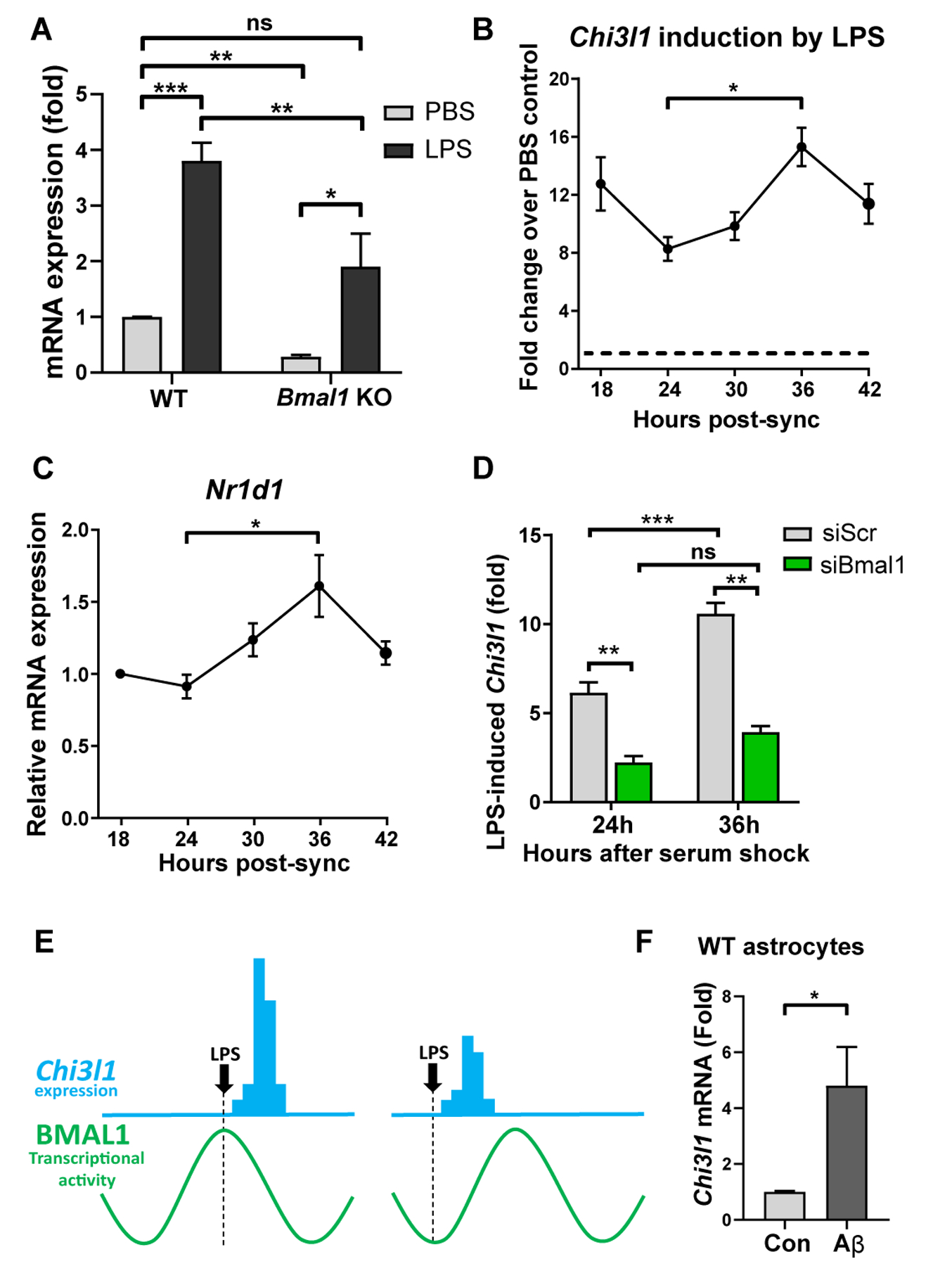
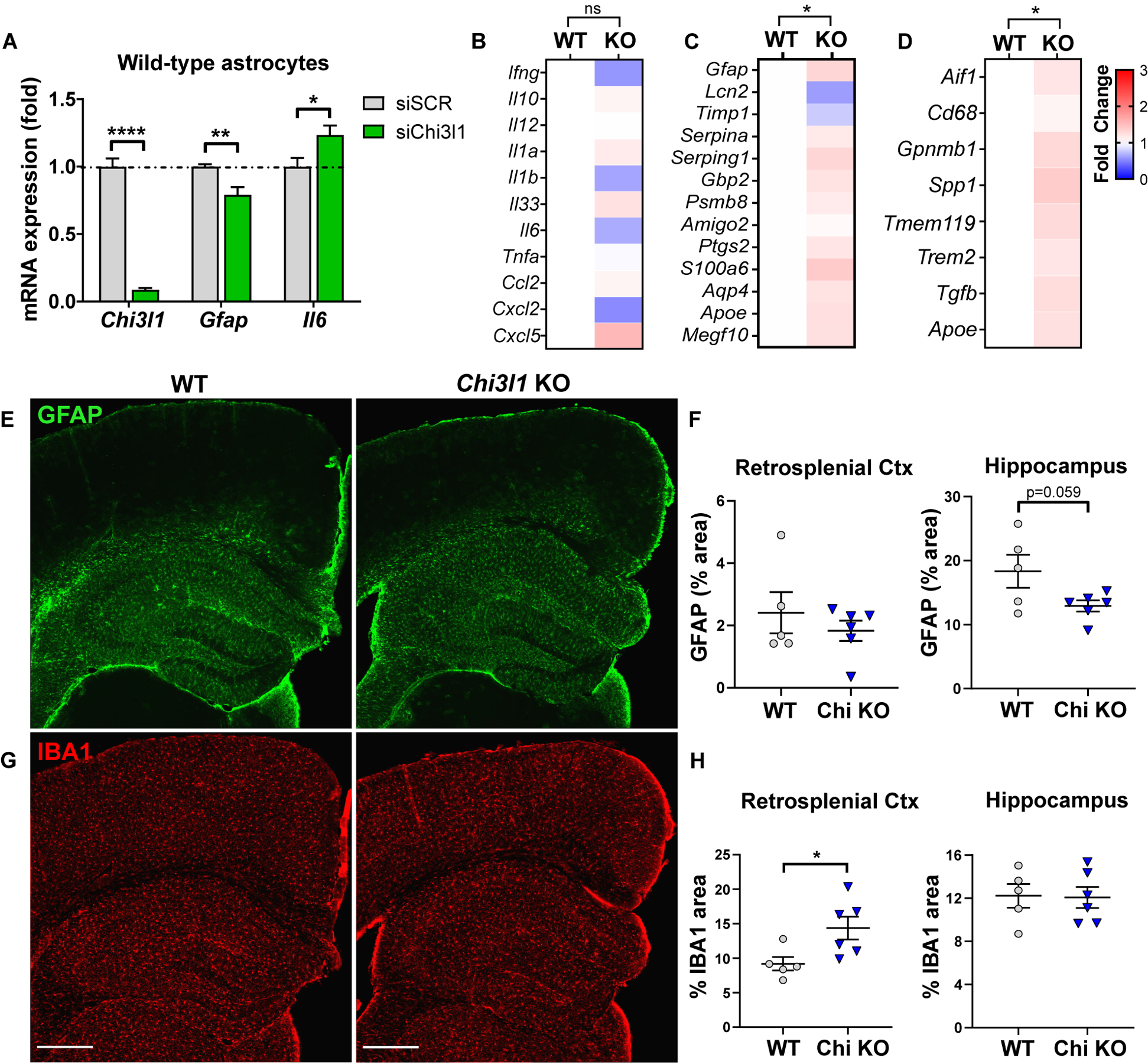
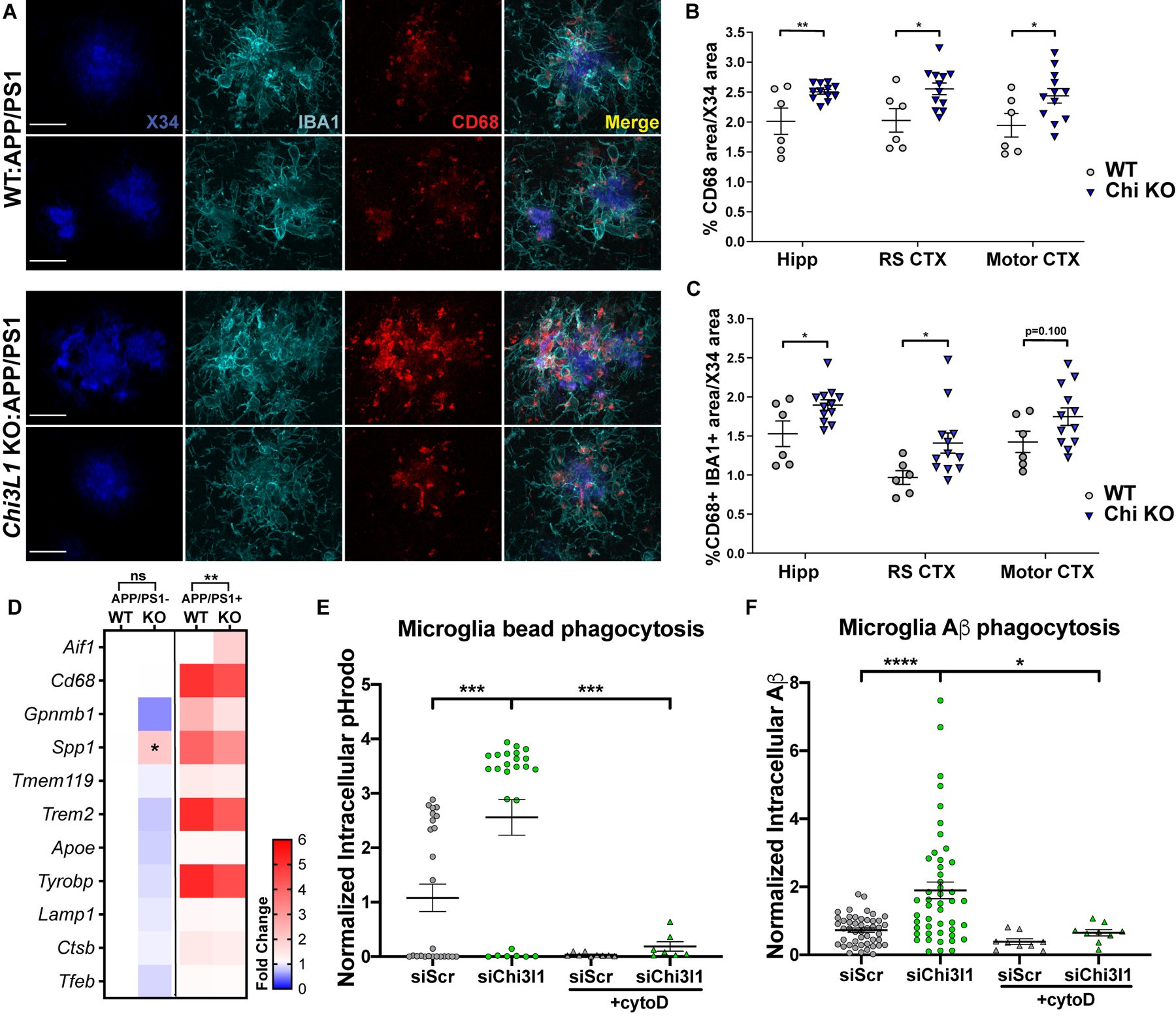
Regulation of glial activation and neuroinflammation are critical factors in the pathogenesis of Alzheimer's disease (AD). YKL-40, a primarily astrocytic protein encoded by the gene , is a widely studied cerebrospinal fluid biomarker that increases with aging and early in AD. However, the function of /YKL-40 in AD is unknown. In a cohort of patients with AD, we observed that a variant in the human gene, which results in decreased CSF YKL-40 expression, was associated with slower AD progression. At baseline, deletion in mice had no effect on astrocyte activation while modestly promoting microglial activation. In a mouse APP/PS1 model of AD, deletion decreased amyloid plaque burden and increased periplaque expression of the microglial lysosomal marker CD68, suggesting that may suppress glial phagocytic activation and promote amyloid accumulation. Accordingly, knockdown increased phagocytosis of zymosan particles and of β-amyloid peptide in both astrocytes and microglia in vitro. We further observed that expression of is regulated by the circadian clock, as deletion of the core clock proteins BMAL1 or CLOCK/NPAS2 strongly suppresses basal expression, whereas deletion of the negative clock regulators PER1/PER2 increased expression. Basal mRNA was nonrhythmic because of a long mRNA half-life in astrocytes. However, inflammatory induction of was gated by the clock. Our findings reveal /YKL-40 as a modulator of glial phagocytic activation and AD pathogenesis in both mice and humans and suggest that the astrocyte circadian clock regulates inflammatory induction.
神经胶质细胞激活和神经炎症的调节是阿尔茨海默病(AD)发病机制中的关键因素。YKL-40是一种主要由该基因编码的星形胶质细胞蛋白,是一种广泛研究的脑脊液生物标志物,其水平随年龄增长和AD早期而升高。然而,YKL-40在AD中的功能尚不清楚。在一组AD患者中,我们观察到人类该基因的一个变体导致脑脊液YKL-40表达降低,与AD进展较慢相关。在基线时,小鼠中的该基因缺失对星形胶质细胞激活没有影响,同时适度促进小胶质细胞激活。在AD的小鼠APP/PS1模型中,该基因缺失减少了淀粉样斑块负荷,并增加了小胶质细胞溶酶体标志物CD68在斑块周围的表达,表明该基因可能抑制神经胶质细胞吞噬激活并促进淀粉样蛋白积累。相应地,该基因敲低增加了体外星形胶质细胞和小胶质细胞中酵母聚糖颗粒和β淀粉样肽的吞噬作用。我们进一步观察到该基因的表达受生物钟调节,因为核心生物钟蛋白BMAL1或CLOCK/NPAS2的缺失强烈抑制基础该基因表达,而负性生物钟调节因子PER1/PER2的缺失增加了该基因表达。由于星形胶质细胞中mRNA半衰期长,基础该基因mRNA无节律性。然而,该基因的炎症诱导受生物钟控制。我们的研究结果揭示YKL-40是小鼠和人类神经胶质细胞吞噬激活和AD发病机制的调节因子,并表明星形胶质细胞生物钟调节炎症该基因诱导。