de Azevedo Ricardo A, Shoshan Einav, Whang Shanzhi, Markel Gal, Jaiswal Ashvin R, Liu Arthur, Curran Michael A, Travassos Luiz R, Bar-Eli Menashe
Department of Cancer Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Experimental Oncology Unit (UNONEX), Department of Microbiology, Immunology and Parasitology, Federal University of São Paulo (UNIFESP), São Paulo, Brazil.
Oncoimmunology. 2020 Dec 6;9(1):1846915. doi: 10.1080/2162402X.2020.1846915.
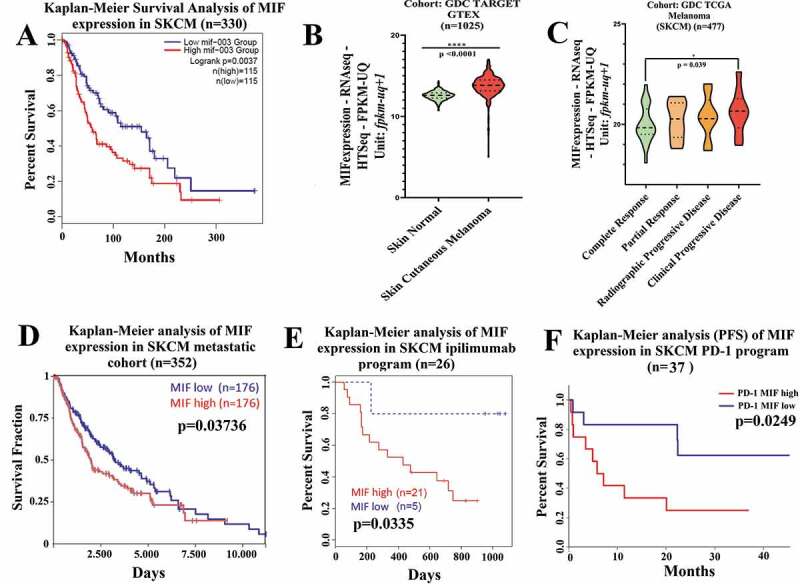
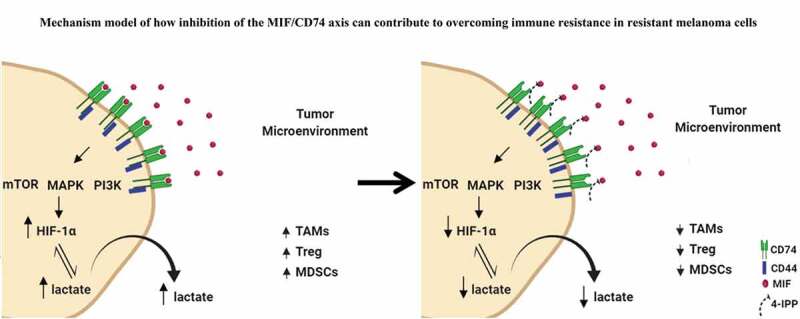
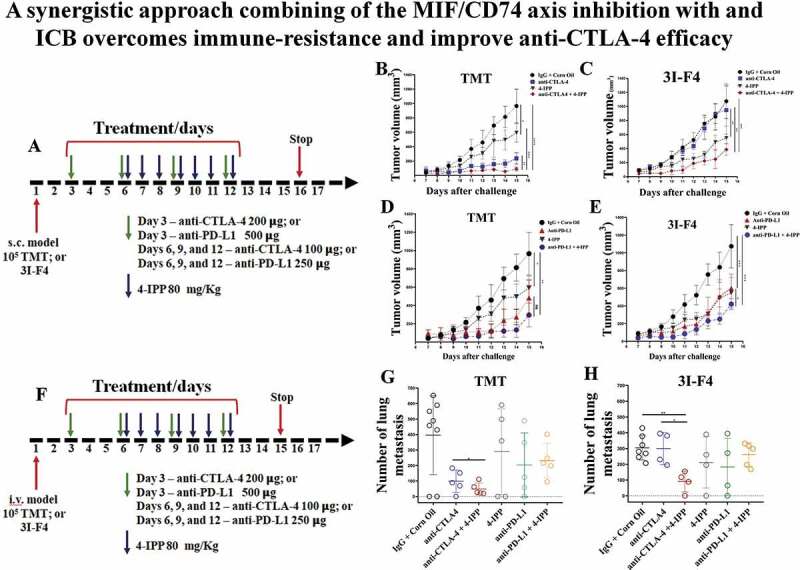
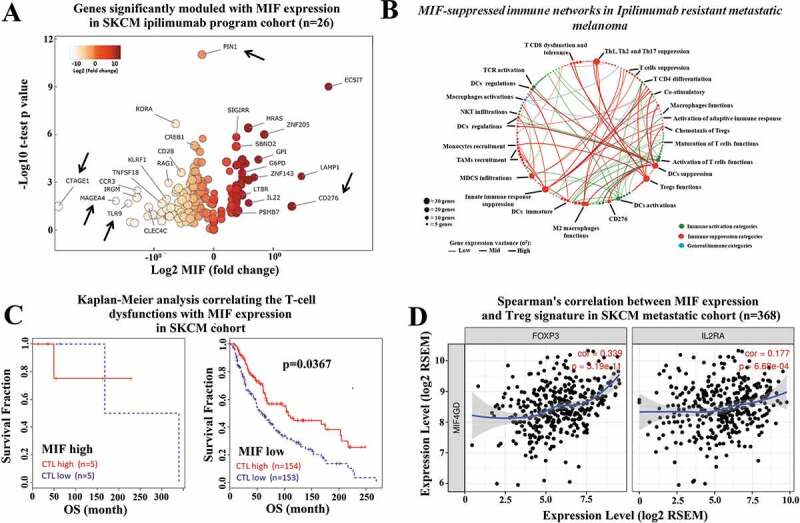
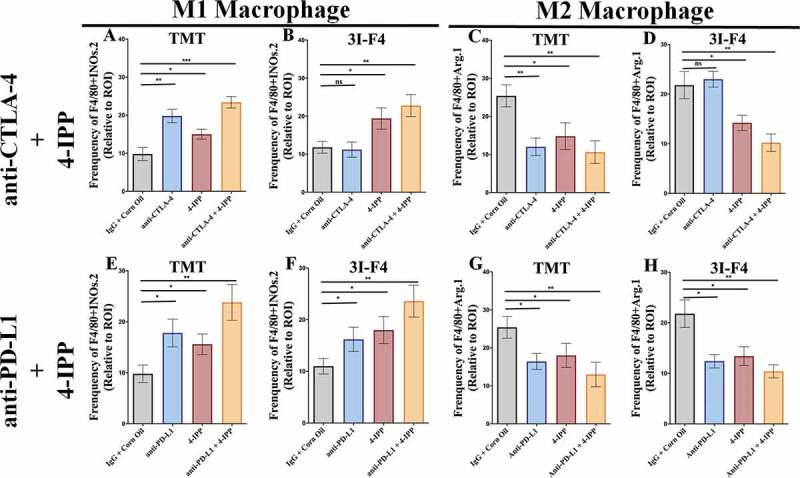
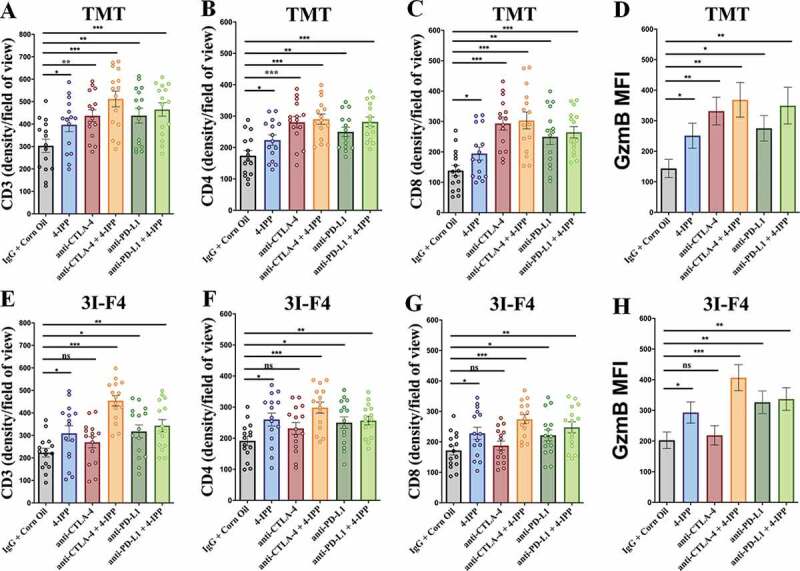
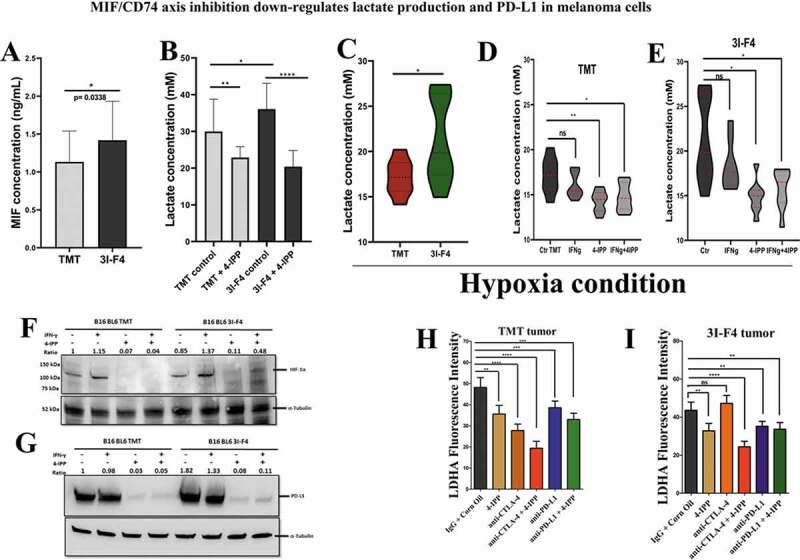
Immune checkpoint blockade (ICB) has demonstrated an impressive outcome in patients with metastatic melanoma, yet, durable complete response; even with Ipilimumab/Nivolumab combo are under 30%. Primary and acquired resistance in response to ICB is commonly due to a tumor immune escape mechanism dictated by the tumor microenvironment (TME). Macrophage Migratory Inhibition Factor (MIF) has emerged as an immunosuppressive factor secreted in the TME. We have previously demonstrated that blockade of the MIF-CD74 signaling on macrophages and dendritic cells restored the anti-tumor immune response against melanoma. Here, we report that inhibition of the MIF-CD74 axis combined with ipilimumab could render resistant melanoma to better respond to anti-CTLA-4 treatment. We provide evidence that blocking the MIF-CD74 signaling potentiates CD8+ T-cells infiltration and drives pro-inflammatory M1 conversion of macrophages in the TME. Furthermore, MIF inhibition resulted in reprogramming the metabolic pathway by reducing lactate production, HIF-1α and PD-L1 expression in the resistant melanoma cells. Melanoma patient data extracted from the TCGA database supports the hypothesis that high MIF expression strongly correlates with poor response to ICB therapy. Our findings provide a rationale for combining anti-CTLA-4 with MIF inhibitors as a potential strategy to overcome resistance to ICB therapy in melanoma, turning a "cold" tumor into a "hot" one mediated by the activation of innate immunity and reprogramming of tumor metabolism and reduced PD-L1 expression in melanoma cells.
免疫检查点阻断(ICB)在转移性黑色素瘤患者中已显示出令人瞩目的疗效,然而,即使使用伊匹单抗/纳武单抗联合治疗,持久完全缓解率仍低于30%。对ICB的原发性和获得性耐药通常归因于肿瘤微环境(TME)所主导的肿瘤免疫逃逸机制。巨噬细胞迁移抑制因子(MIF)已成为TME中分泌的一种免疫抑制因子。我们之前已经证明,阻断巨噬细胞和树突状细胞上的MIF-CD74信号可恢复针对黑色素瘤的抗肿瘤免疫反应。在此,我们报告,抑制MIF-CD74轴联合伊匹单抗可使耐药黑色素瘤对抗CTLA-4治疗产生更好的反应。我们提供的证据表明,阻断MIF-CD74信号可增强CD8+ T细胞浸润,并促使TME中的巨噬细胞向促炎性M1型转化。此外,MIF抑制导致耐药黑色素瘤细胞的代谢途径重编程,减少乳酸生成、HIF-1α和PD-L1表达。从TCGA数据库提取的黑色素瘤患者数据支持以下假设:高MIF表达与对ICB治疗的不良反应密切相关。我们的研究结果为将抗CTLA-4与MIF抑制剂联合使用提供了理论依据,这是一种克服黑色素瘤对ICB治疗耐药的潜在策略,可将“冷”肿瘤转变为通过激活先天免疫、重编程肿瘤代谢以及降低黑色素瘤细胞中PD-L1表达介导的“热”肿瘤。