US Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
Department of Genetics, Evolution, Microbiology and Immunology, Institute of Biology, University of Campinas, Campinas, Brazil.
Nat Biotechnol. 2021 May;39(5):578-585. doi: 10.1038/s41587-020-00774-7. Epub 2020 Dec 21.
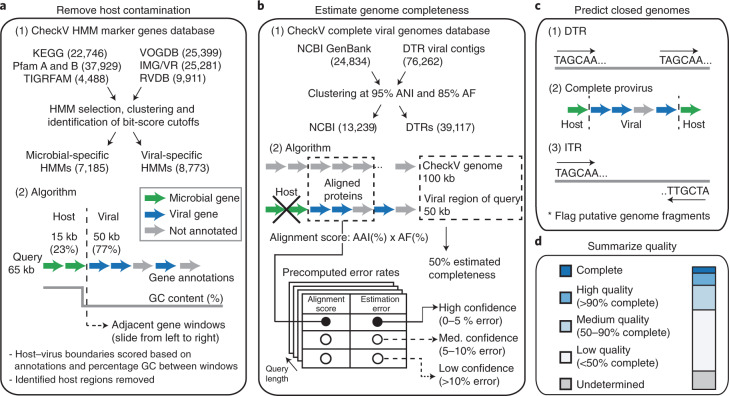
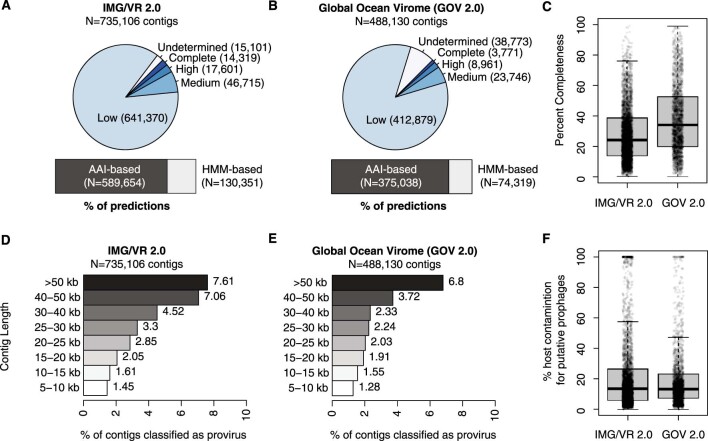
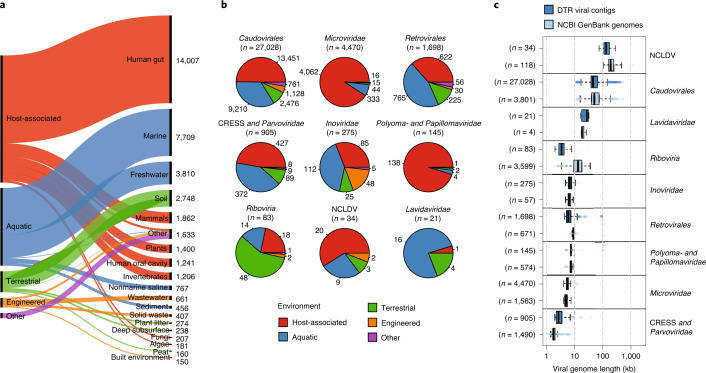
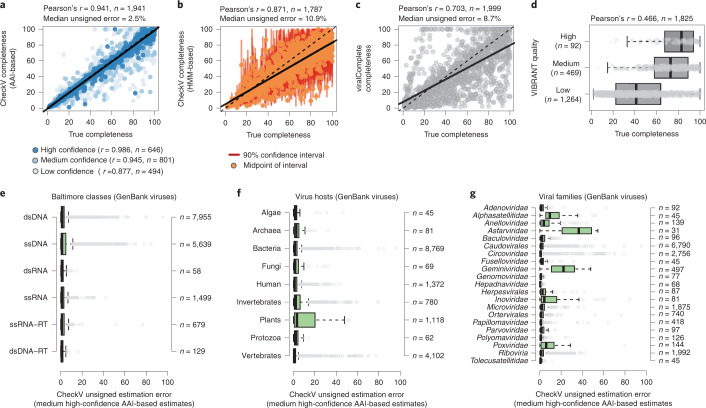
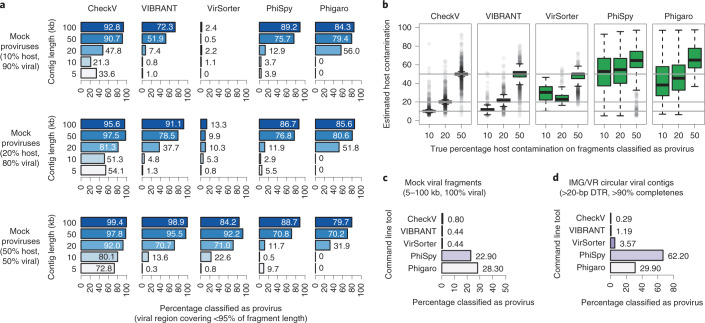
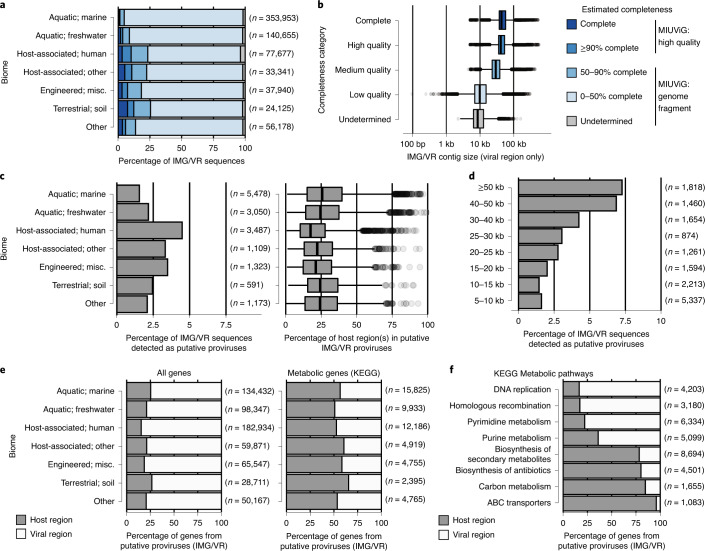
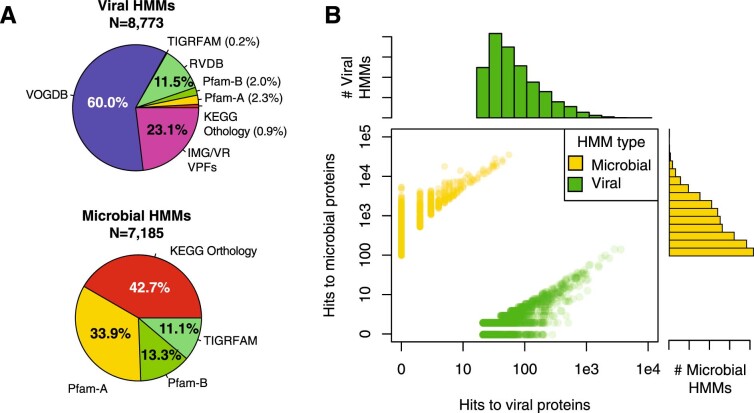
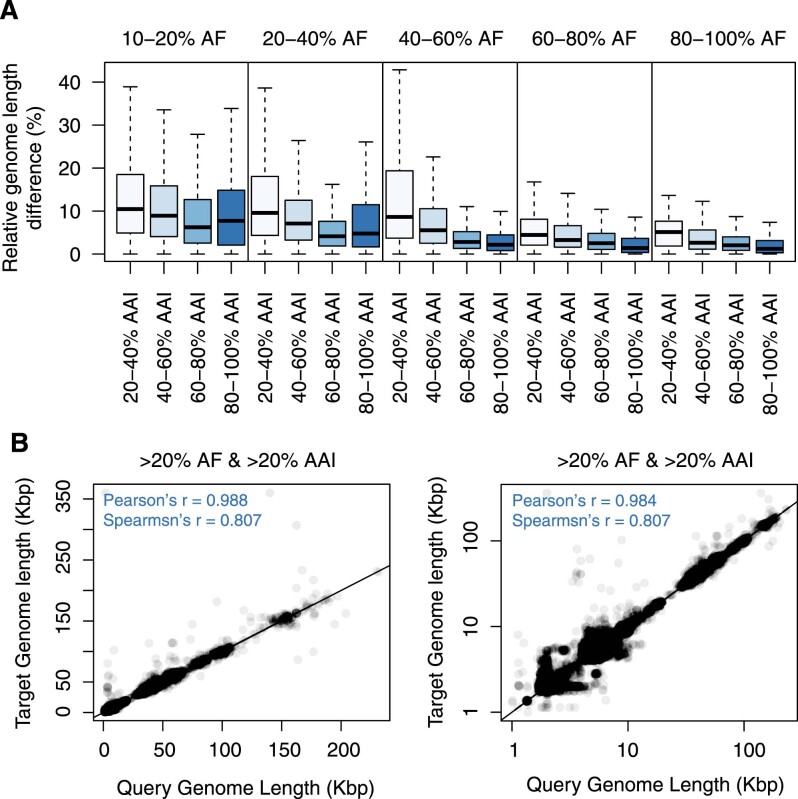
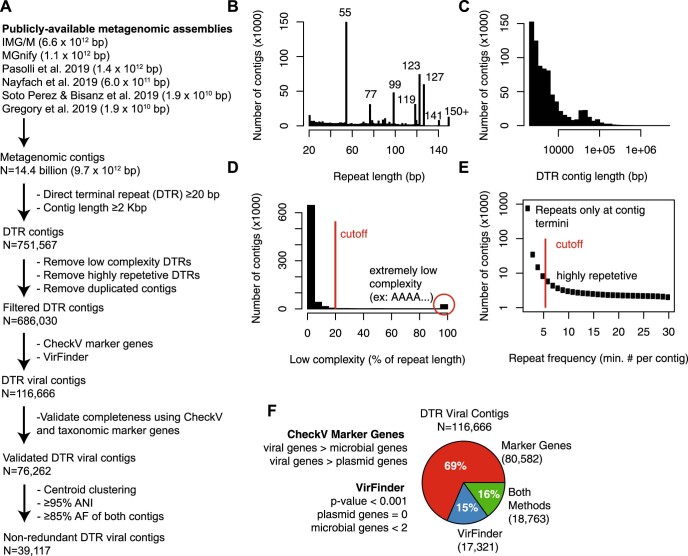
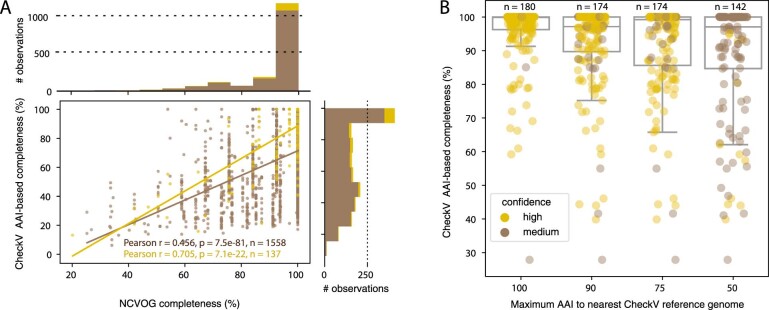
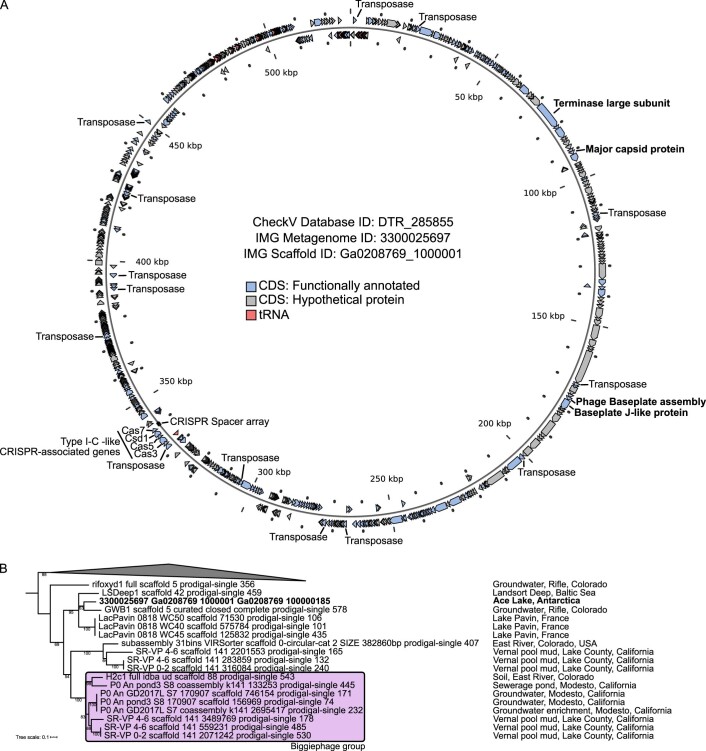
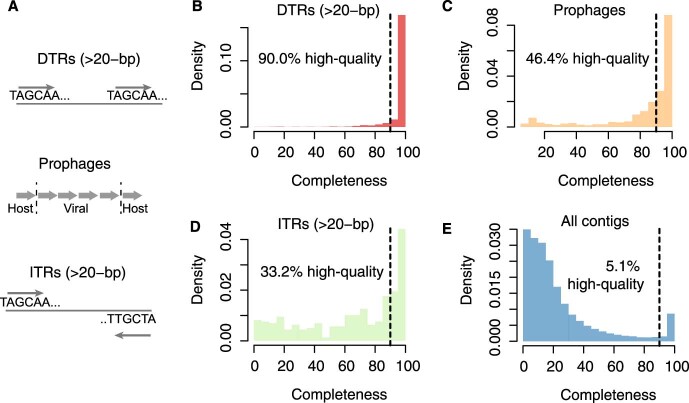
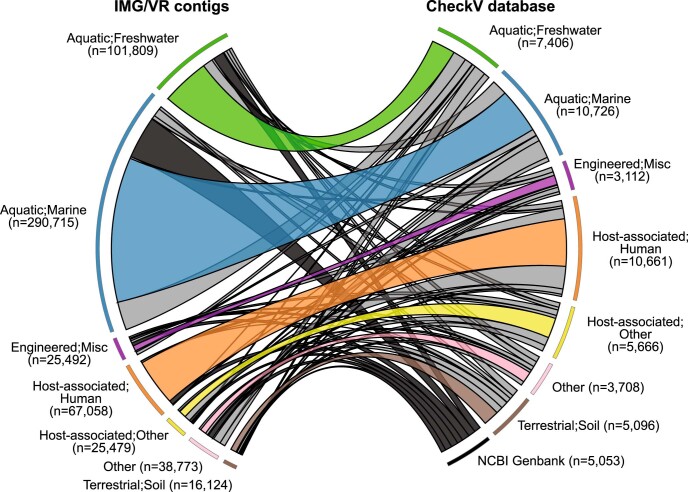
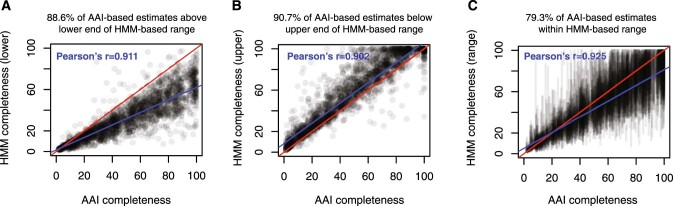
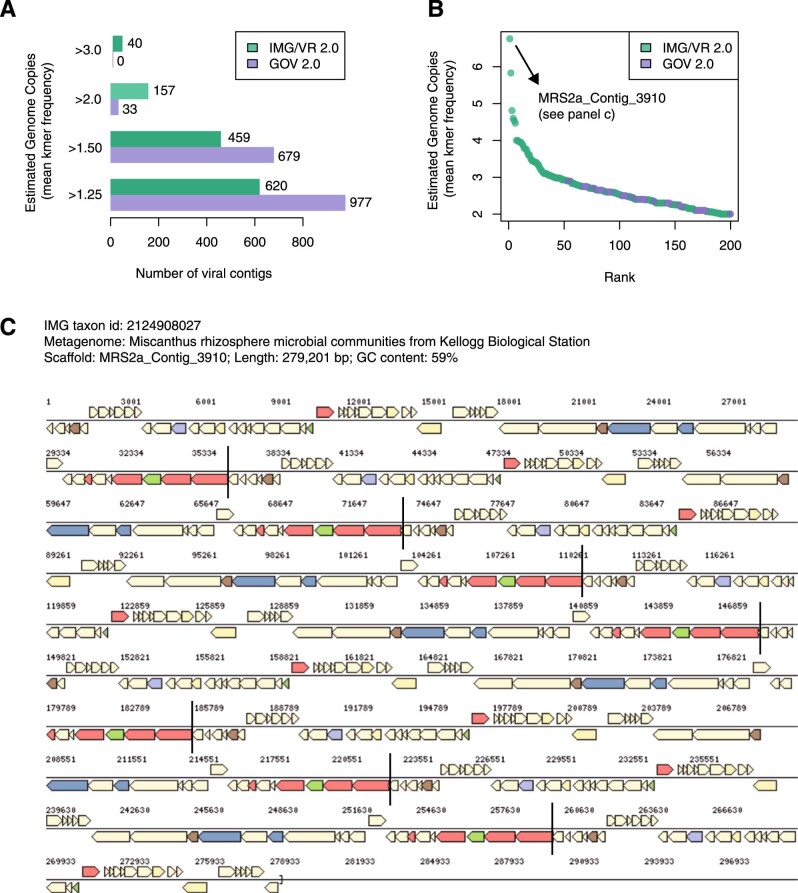
Millions of new viral sequences have been identified from metagenomes, but the quality and completeness of these sequences vary considerably. Here we present CheckV, an automated pipeline for identifying closed viral genomes, estimating the completeness of genome fragments and removing flanking host regions from integrated proviruses. CheckV estimates completeness by comparing sequences with a large database of complete viral genomes, including 76,262 identified from a systematic search of publicly available metagenomes, metatranscriptomes and metaviromes. After validation on mock datasets and comparison to existing methods, we applied CheckV to large and diverse collections of metagenome-assembled viral sequences, including IMG/VR and the Global Ocean Virome. This revealed 44,652 high-quality viral genomes (that is, >90% complete), although the vast majority of sequences were small fragments, which highlights the challenge of assembling viral genomes from short-read metagenomes. Additionally, we found that removal of host contamination substantially improved the accurate identification of auxiliary metabolic genes and interpretation of viral-encoded functions.
从宏基因组中已经鉴定出了数以百万计的新病毒序列,但这些序列的质量和完整性差异很大。在这里,我们介绍了 CheckV,这是一个用于识别封闭病毒基因组、估计基因组片段完整性并从整合前病毒中去除侧翼宿主区域的自动化管道。CheckV 通过将序列与一个包含大量完整病毒基因组的数据库进行比较来估计完整性,其中包括从系统搜索公开可用的宏基因组、宏转录组和宏病毒组中鉴定出的 76,262 个基因组。在对模拟数据集进行验证并与现有方法进行比较后,我们将 CheckV 应用于包括 IMG/VR 和全球海洋病毒组在内的大量多样的宏基因组组装病毒序列集合。这揭示了 44,652 个高质量的病毒基因组(即>90%完整),尽管绝大多数序列都是小片段,这突出了从短读长宏基因组组装病毒基因组的挑战。此外,我们发现去除宿主污染可以显著提高辅助代谢基因的准确识别和病毒编码功能的解释。