Fu Laiyi, Zhang Lihua, Dollinger Emmanuel, Peng Qinke, Nie Qing, Xie Xiaohui
Systems Engineering Institute, School of Electronic and Information Engineering, Xi'an Jiaotong University, Xi'an, Shannxi 710049, China.
Department of Computer Science, University of California, Irvine, Irvine, CA 92697, USA.
Sci Adv. 2020 Dec 18;6(51). doi: 10.1126/sciadv.aba9031. Print 2020 Dec.
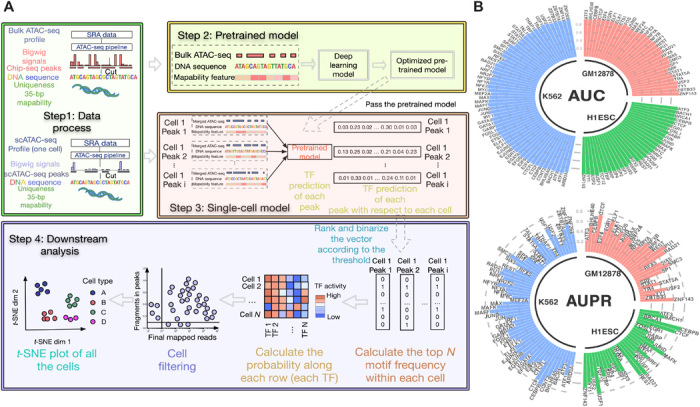
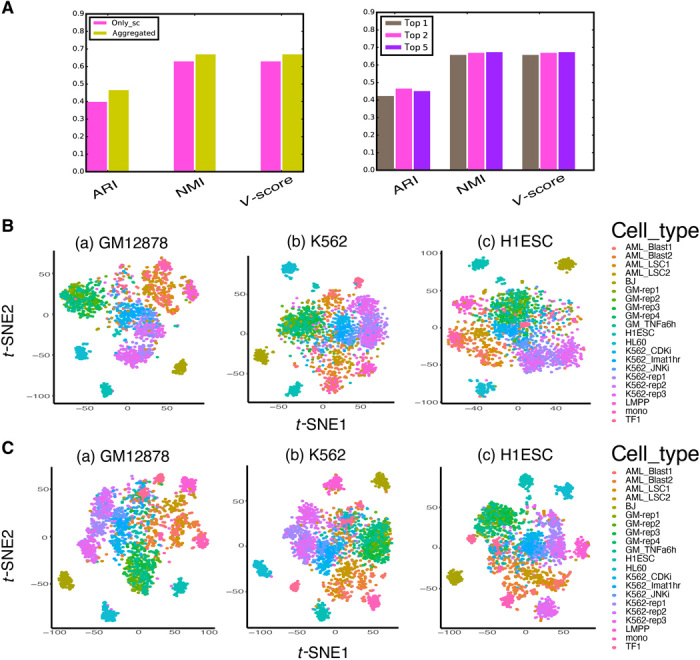
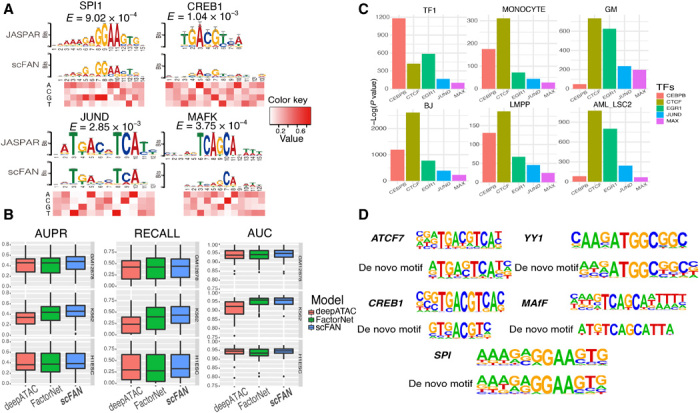
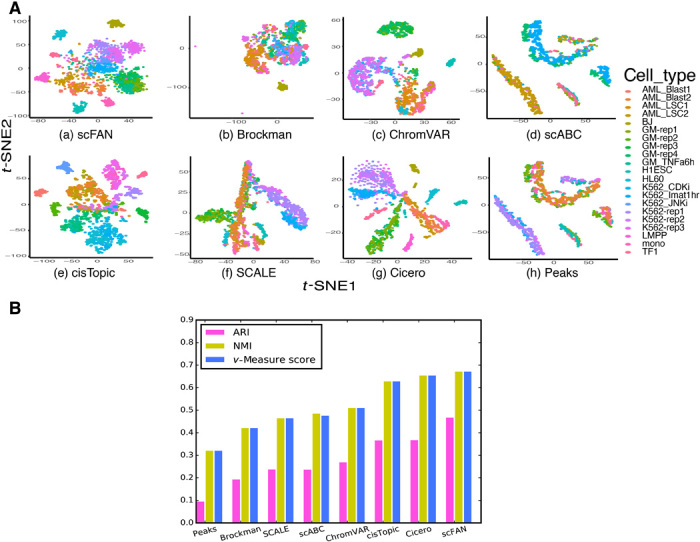
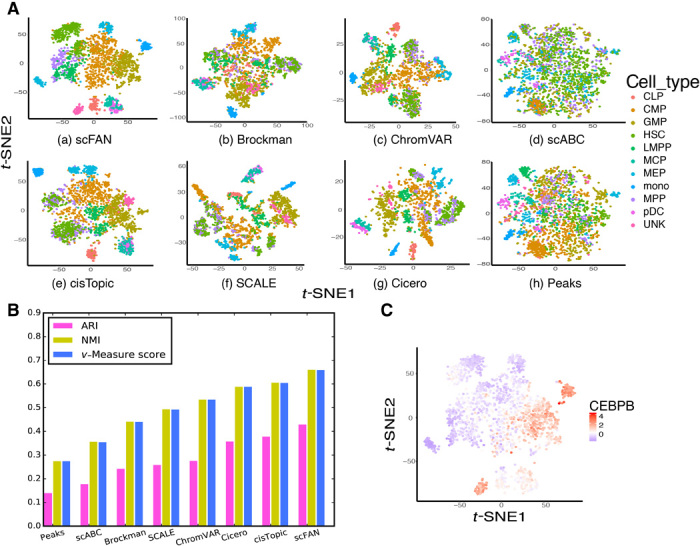
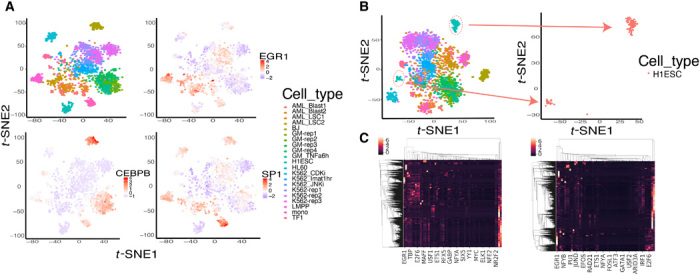
Characterizing genome-wide binding profiles of transcription factors (TFs) is essential for understanding biological processes. Although techniques have been developed to assess binding profiles within a population of cells, determining them at a single-cell level remains elusive. Here, we report scFAN (single-cell factor analysis network), a deep learning model that predicts genome-wide TF binding profiles in individual cells. scFAN is pretrained on genome-wide bulk assay for transposase-accessible chromatin sequencing (ATAC-seq), DNA sequence, and chromatin immunoprecipitation sequencing (ChIP-seq) data and uses single-cell ATAC-seq to predict TF binding in individual cells. We demonstrate the efficacy of scFAN by both studying sequence motifs enriched within predicted binding peaks and using predicted TFs for discovering cell types. We develop a new metric "TF activity score" to characterize each cell and show that activity scores can reliably capture cell identities. scFAN allows us to discover and study cellular identities and heterogeneity based on chromatin accessibility profiles.
表征转录因子(TFs)的全基因组结合图谱对于理解生物学过程至关重要。尽管已经开发出技术来评估细胞群体内的结合图谱,但在单细胞水平上确定它们仍然难以实现。在这里,我们报告了scFAN(单细胞因子分析网络),这是一种深度学习模型,可预测单个细胞中的全基因组TF结合图谱。scFAN在全基因组转座酶可及染色质测序(ATAC-seq)、DNA序列和染色质免疫沉淀测序(ChIP-seq)数据的批量测定上进行预训练,并使用单细胞ATAC-seq来预测单个细胞中的TF结合。我们通过研究预测结合峰内富集的序列基序以及使用预测的TF来发现细胞类型,证明了scFAN的有效性。我们开发了一种新的指标“TF活性评分”来表征每个细胞,并表明活性评分可以可靠地捕捉细胞身份。scFAN使我们能够基于染色质可及性图谱发现和研究细胞身份及异质性。