Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA.
Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, 02140, USA.
BMC Bioinformatics. 2018 Jul 3;19(1):253. doi: 10.1186/s12859-018-2255-6.
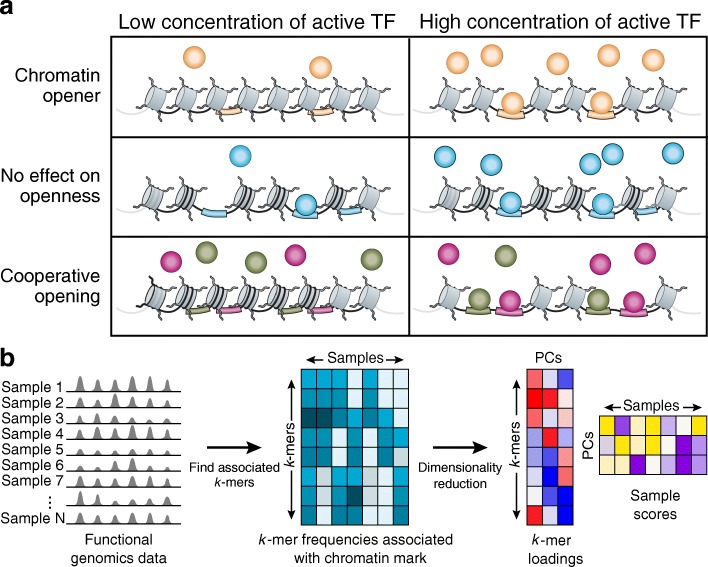
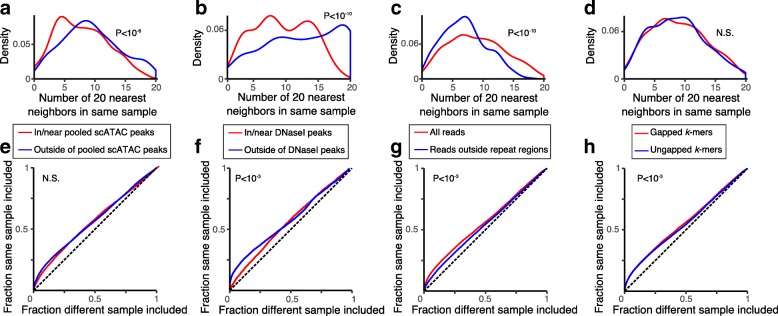
Variation in chromatin organization across single cells can help shed important light on the mechanisms controlling gene expression, but scale, noise, and sparsity pose significant challenges for interpretation of single cell chromatin data. Here, we develop BROCKMAN (Brockman Representation Of Chromatin by K-mers in Mark-Associated Nucleotides), an approach to infer variation in transcription factor (TF) activity across samples through unsupervised analysis of the variation in DNA sequences associated with an epigenomic mark.
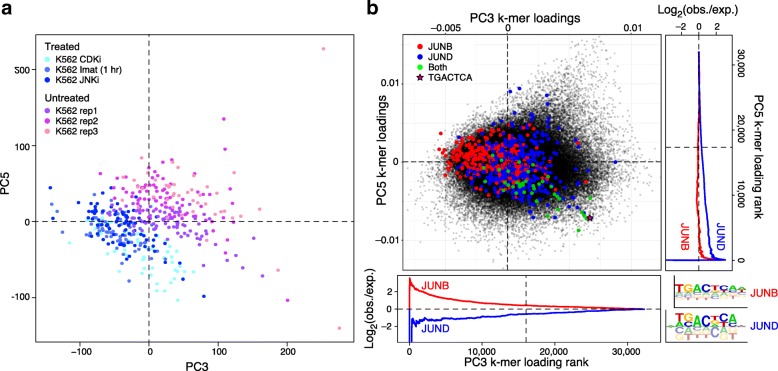
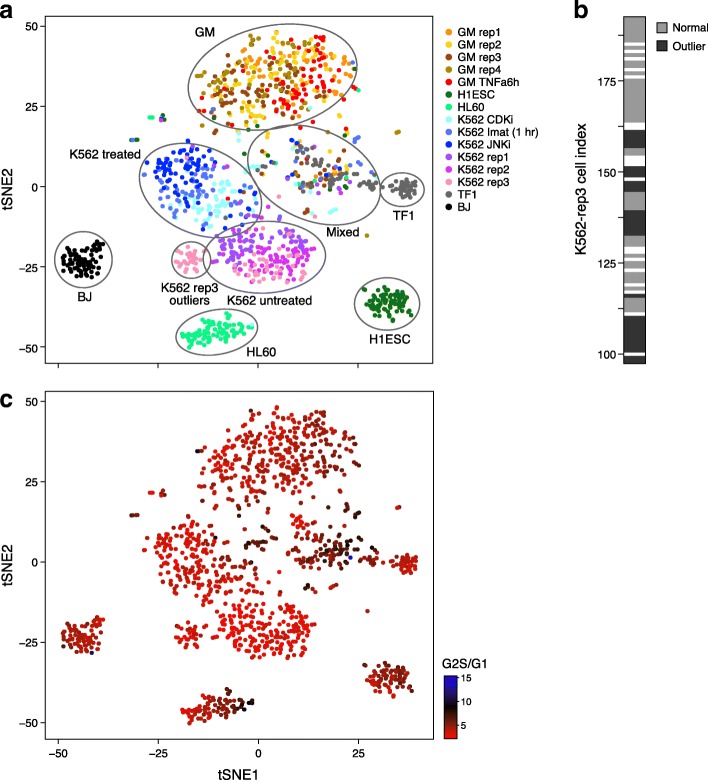
BROCKMAN represents each sample as a vector of epigenomic-mark-associated DNA word frequencies, and decomposes the resulting matrix to find hidden structure in the data, followed by unsupervised grouping of samples and identification of the TFs that distinguish groups. Applied to single cell ATAC-seq, BROCKMAN readily distinguished cell types, treatments, batch effects, experimental artifacts, and cycling cells. We show that each variable component in the k-mer landscape reflects a set of co-varying TFs, which are often known to physically interact. For example, in K562 cells, AP-1 TFs were central determinant of variability in chromatin accessibility through their variable expression levels and diverse interactions with other TFs. We provide a theoretical basis for why cooperative TF binding - and any associated epigenomic mark - is inherently more variable than non-cooperative binding.
BROCKMAN and related approaches will help gain a mechanistic understanding of the trans determinants of chromatin variability between cells, treatments, and individuals.
单细胞中染色质组织的变化可以帮助我们深入了解控制基因表达的机制,但规模、噪声和稀疏性给单细胞染色质数据的解释带来了重大挑战。在这里,我们开发了 BROCKMAN(通过标记相关核苷酸中的 K- -mer 对染色质的 Brockman 表示),这是一种通过对与表观遗传标记相关的 DNA 序列的变化进行无监督分析来推断转录因子 (TF) 活性在样本间变化的方法。
BROCKMAN 将每个样本表示为与表观遗传标记相关的 DNA 字频向量,并对得到的矩阵进行分解,以找到数据中的隐藏结构,然后对样本进行无监督分组,并识别区分组的 TF。将 BROCKMAN 应用于单细胞 ATAC-seq,可轻松区分细胞类型、处理、批次效应、实验伪影和细胞周期。我们表明,k- -mer 景观中的每个变量成分都反映了一组共同变化的 TF,这些 TF 通常已知具有物理相互作用。例如,在 K562 细胞中,AP-1 TF 是染色质可及性变异性的主要决定因素,这是通过它们可变的表达水平和与其他 TF 的多样化相互作用实现的。我们为为什么合作 TF 结合——以及任何相关的表观遗传标记——本质上比非合作结合更具变异性提供了理论基础。
BROCKMAN 及相关方法将有助于深入了解细胞、处理和个体之间染色质变异性的转录决定因素的机制。