Tam Benjamin, Sinha Siddharth, Wang San Ming
Cancer Centre and Institute of Translational Medicine, Faculty of Health Sciences, University of Macau, Macau.
Comput Struct Biotechnol J. 2020 Dec 2;18:4033-4039. doi: 10.1016/j.csbj.2020.11.041. eCollection 2020.
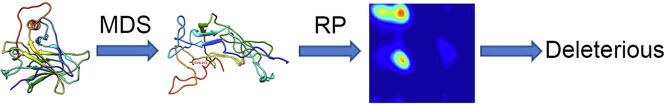
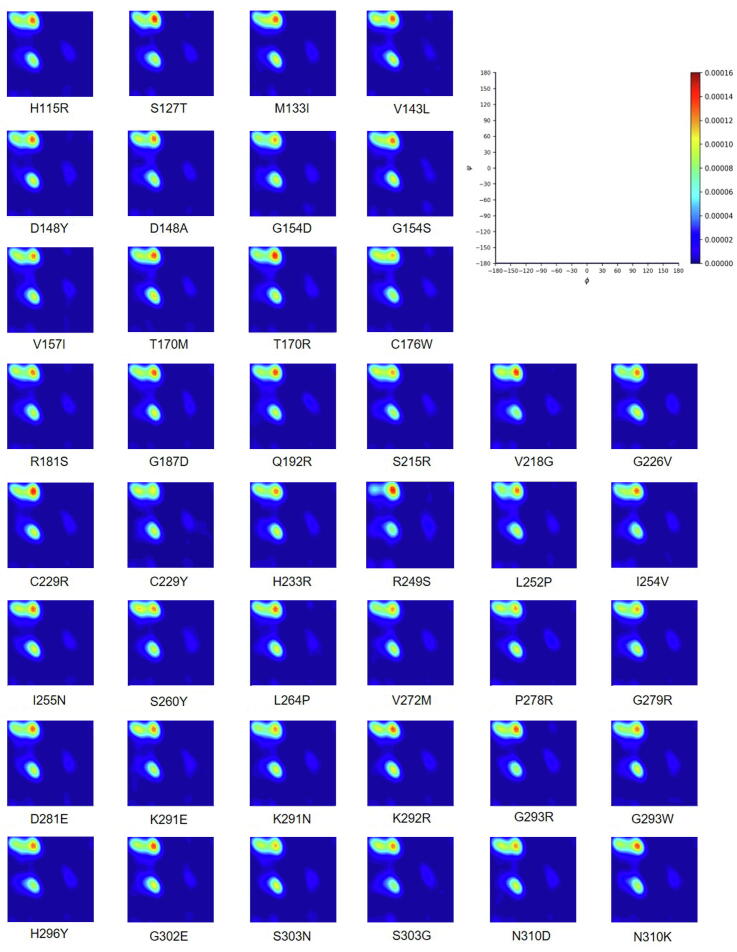
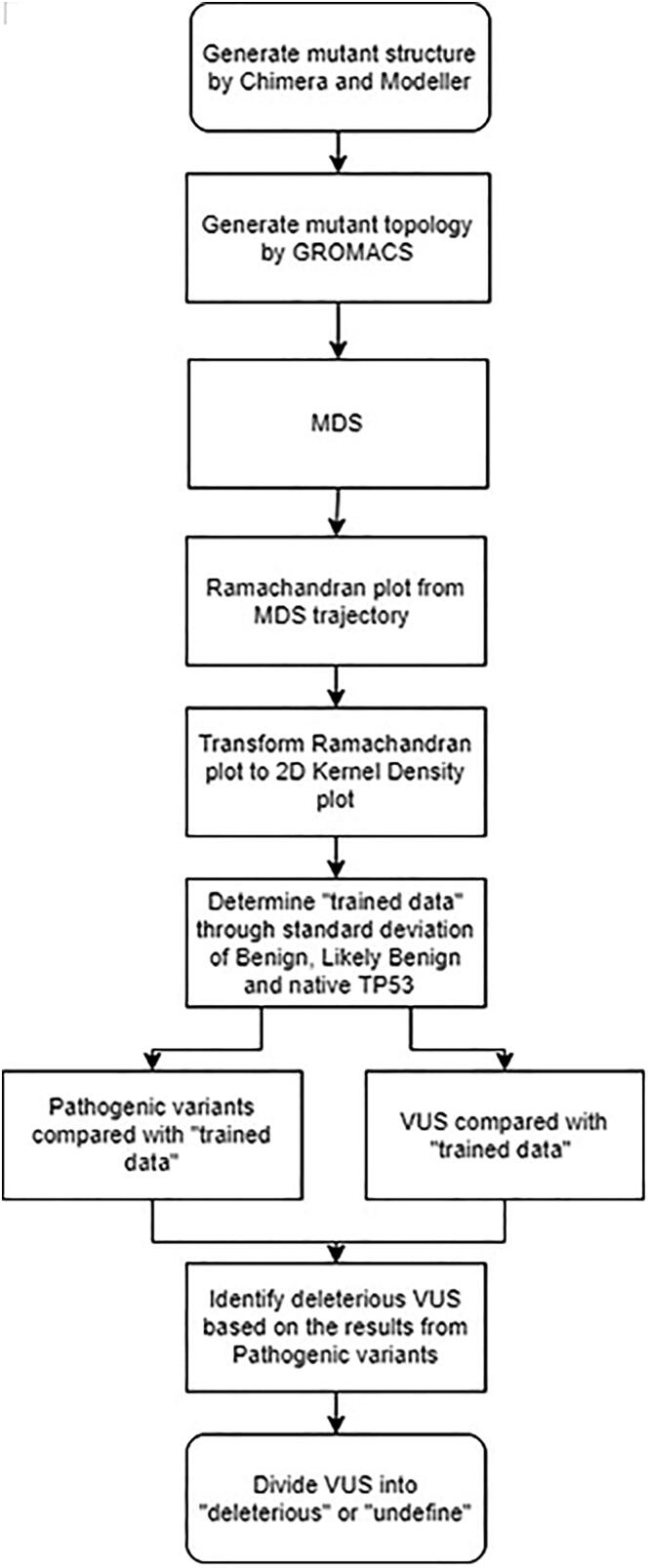
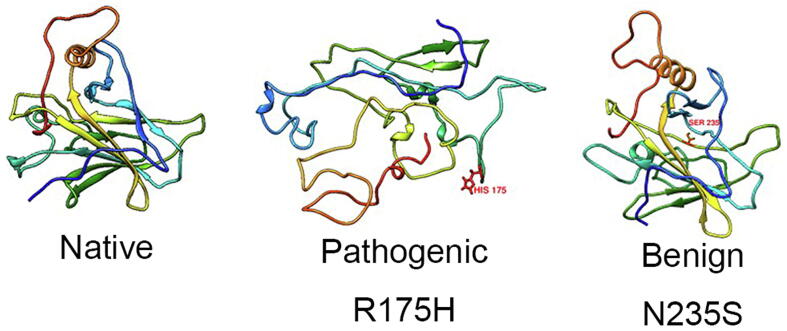
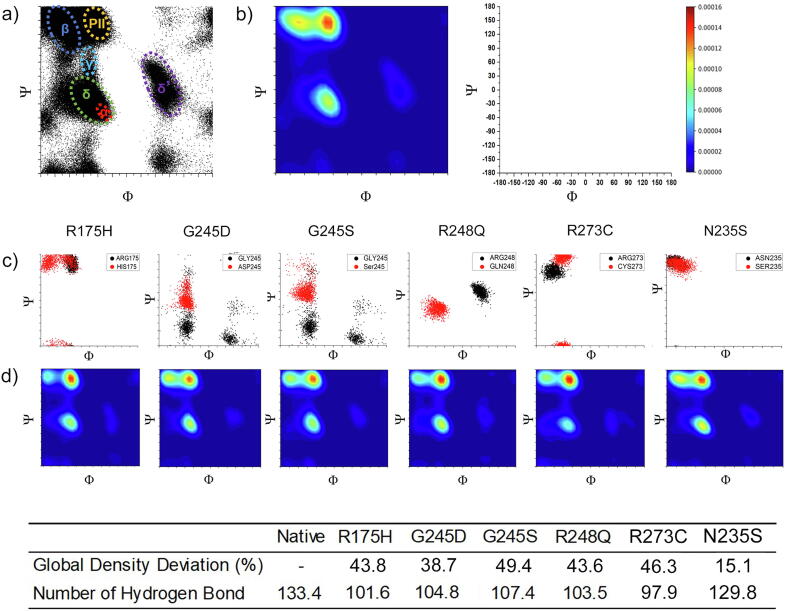
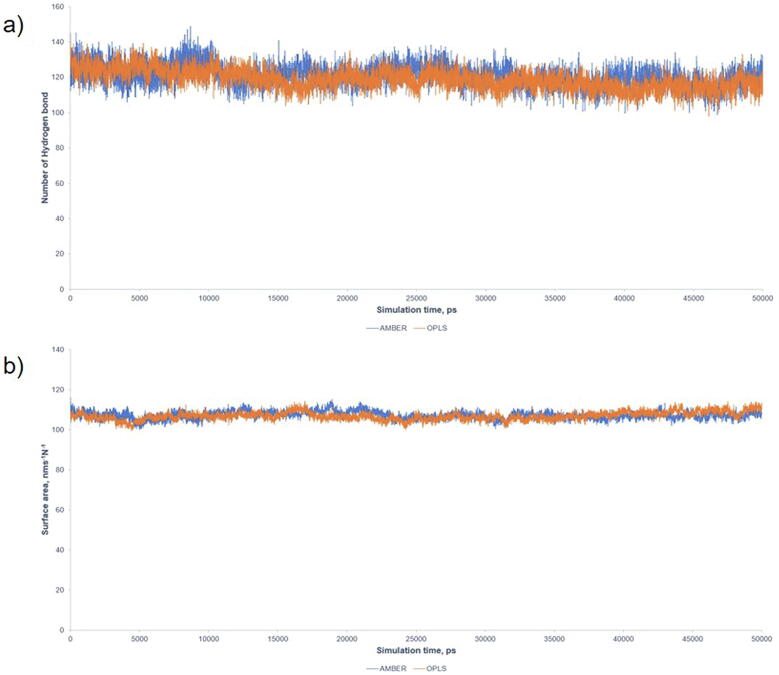
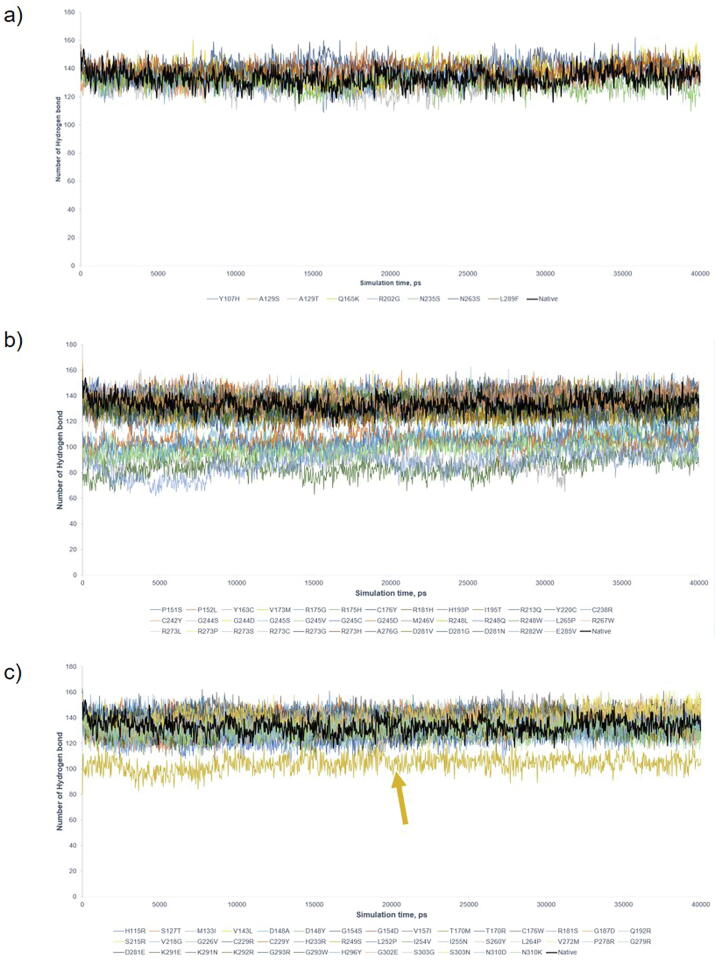
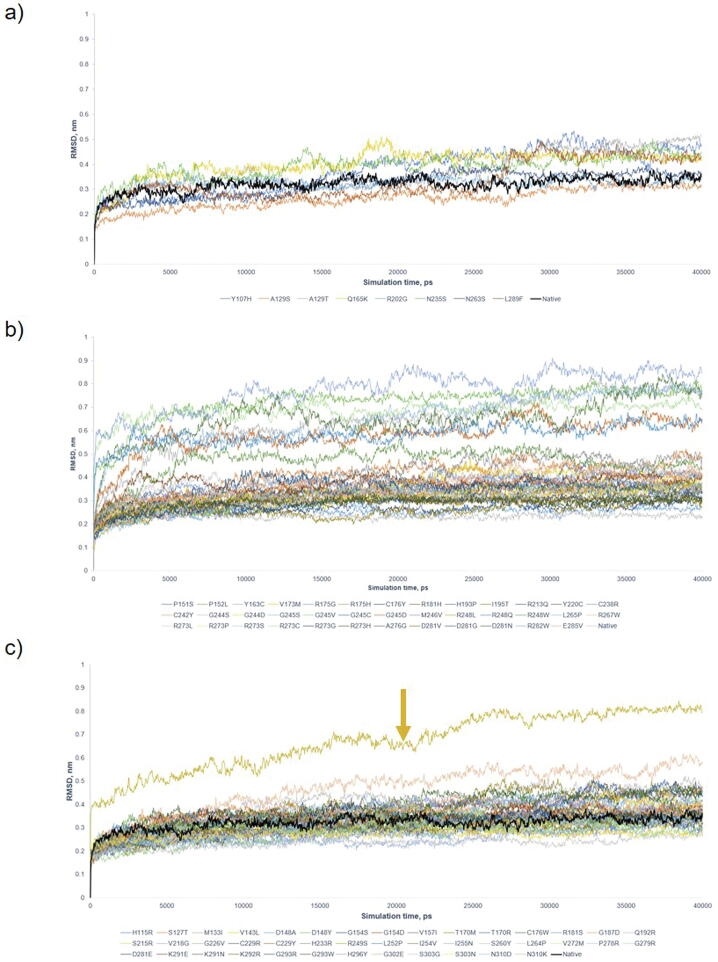
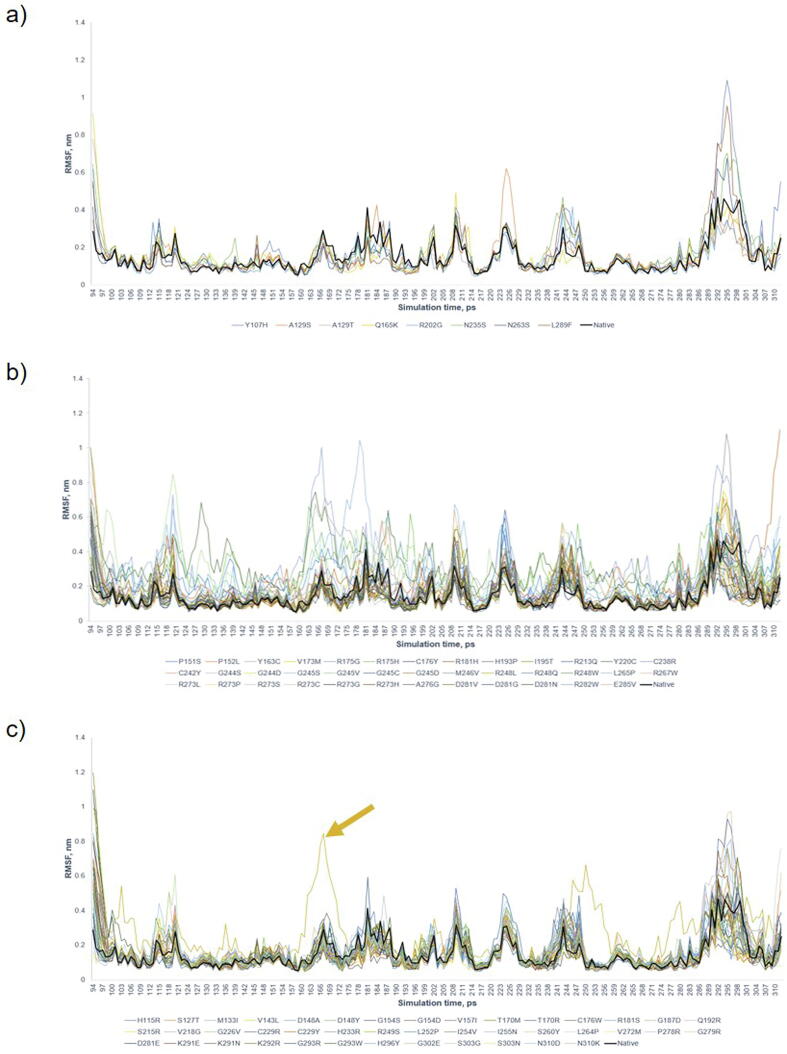
The wide application of new DNA sequencing technologies is generating vast quantities of genetic variation data at unprecedented speed. Developing methodologies to decode the pathogenicity of the variants is imperatively demanding. We hypothesized that as deleterious variants may function through disturbing structural stability of their affected proteins, information from structural change caused by genetic variants can be used to identify the variants with deleterious effects. In order to measure the structural change for proteins with large size, we designed a method named RP-MDS composed of Ramachandran plot (RP) and Molecular Dynamics Simulation (MDS). Ramachandran plot captures the variant-caused secondary structural change, whereas MDS provides a quantitative measure for the variant-caused globular structural change. We tested the method using variants in DNA binding domain of 219 residues as the model. In total, RP-MDS identified 23 of 38 (60.5%) known Pathogenic variants and 17 of 42 (41%) VUS that caused significant changes of P53 structure. Our study demonstrates that RP-MDS method provides a powerful protein structure-based tool to screen deleterious genetic variants affecting large-size proteins.
新的DNA测序技术的广泛应用正以前所未有的速度产生大量的遗传变异数据。开发解码变异致病性的方法迫在眉睫。我们假设,由于有害变异可能通过干扰其受影响蛋白质的结构稳定性来发挥作用,来自基因变异引起的结构变化的信息可用于识别具有有害影响的变异。为了测量大尺寸蛋白质的结构变化,我们设计了一种名为RP-MDS的方法,该方法由拉氏图(RP)和分子动力学模拟(MDS)组成。拉氏图捕捉变异引起的二级结构变化,而MDS为变异引起的球状结构变化提供定量测量。我们以219个残基的DNA结合域中的变异作为模型测试了该方法。RP-MDS总共识别出38个已知致病变异中的23个(60.5%)以及42个导致P53结构显著变化的意义未明变异(VUS)中的17个(41%)。我们的研究表明,RP-MDS方法提供了一种强大的基于蛋白质结构的工具,用于筛选影响大尺寸蛋白质的有害遗传变异。