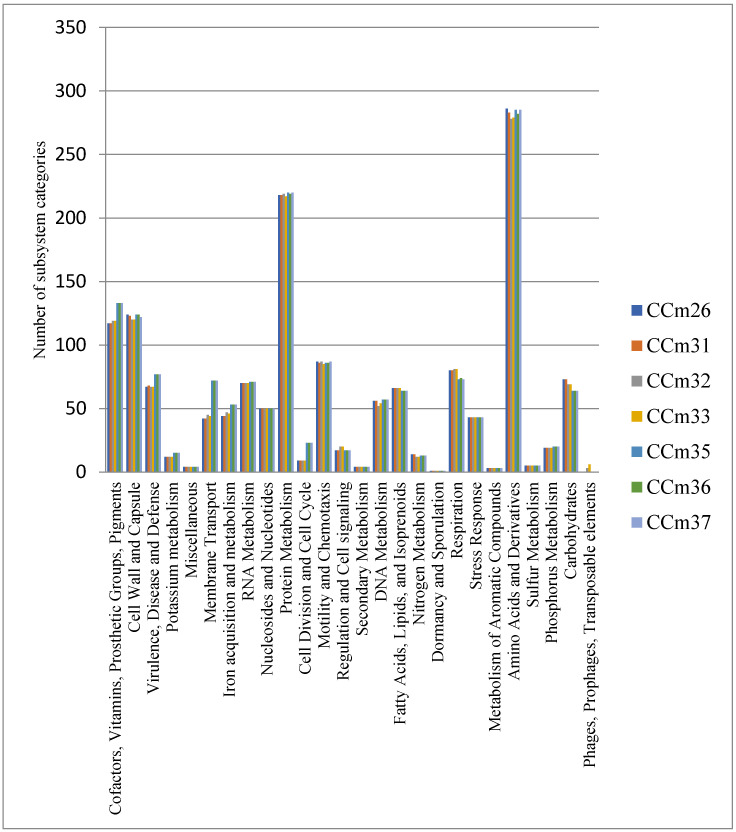
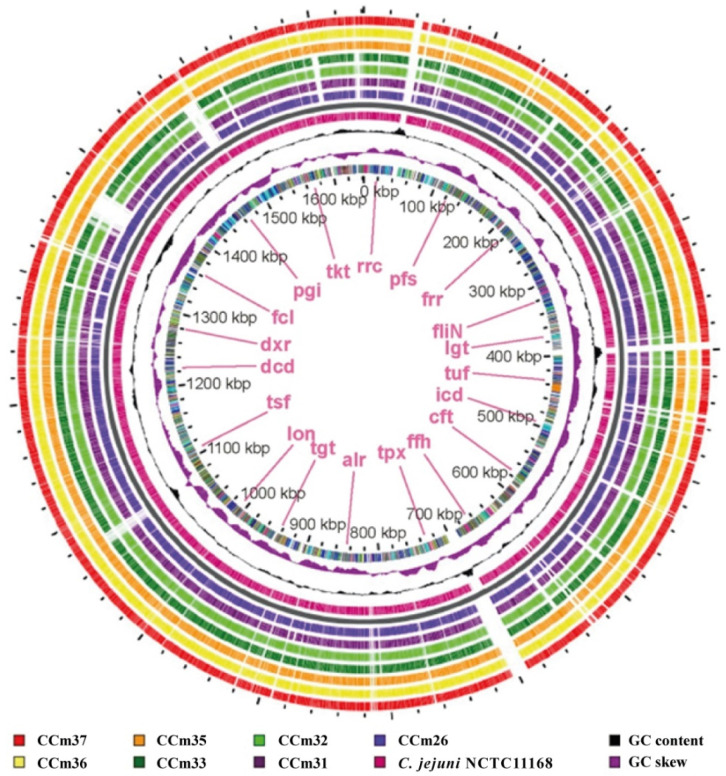
Aksomaitiene Jurgita, Novoslavskij Aleksandr, Kudirkiene Egle, Gabinaitiene Ausra, Malakauskas Mindaugas
Department of Food Safety and Quality, Faculty of Veterinary Medicine, Veterinary Academy, Lithuanian University of Health Sciences, Tilzes str. 18, LT 47181 Kaunas, Lithuania.
Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Stigbøjlen 4, 1870 Frederiksberg C, Denmark.
Microorganisms. 2020 Dec 29;9(1):66. doi: 10.3390/microorganisms9010066.
Spread of antibiotic resistance via mobile genetic elements associates with transfer of genes providing resistance against multiple antibiotics. Use of various comparative genomics analysis techniques enables to find intrinsic and acquired genes associated with phenotypic antimicrobial resistance (AMR) in genome sequences with exceptionally high-level multidrug resistance. In this study, we used whole genome sequences of seven to identify isolate-specific genomic features associated with resistance and virulence determinants and their role in multidrug resistance (MDR). All isolates were phenotypically highly resistant to tetracycline, ciprofloxacin, and ceftriaxone (MIC range from 64 to ≥256 µg/mL). Besides, two isolates were resistant to gentamicin, and one was resistant to erythromycin. The extensive drug-resistance profiles were confirmed for the two isolates assigned to ST-4447 (CC179). The most occurring genetic antimicrobial-resistance determinants were , beta-lactamase, and multidrug efflux pumps. In this study, mobile genetic elements (MGEs) were detected in genomic islands carrying genes that confer resistance to MDR, underline their importance for disseminating antibiotic resistance in . The genomic approach showed a diverse distribution of virulence markers, including both plasmids and phage sequences that serve as horizontal gene transfer tools. The study findings describe in silico prediction of AMR and virulence genetics determinants combined with phenotypic AMR detection in multidrug-resistant isolates from Lithuania.
抗生素耐药性通过移动遗传元件传播与提供对多种抗生素耐药性的基因转移相关。使用各种比较基因组学分析技术能够在具有极高水平多药耐药性的基因组序列中找到与表型抗菌药物耐药性(AMR)相关的固有基因和获得性基因。在本研究中,我们使用了七个的全基因组序列来鉴定与耐药性和毒力决定因素相关的菌株特异性基因组特征及其在多药耐药性(MDR)中的作用。所有分离株在表型上对四环素、环丙沙星和头孢曲松高度耐药(MIC范围为64至≥256μg/mL)。此外,两个分离株对庆大霉素耐药,一个对红霉素耐药。对于分配到ST-4447(CC179)的两个分离株,广泛的耐药谱得到了证实。最常见的遗传抗菌药物耐药决定因素是、β-内酰胺酶和多药外排泵。在本研究中,在携带赋予对MDR耐药性基因的基因组岛中检测到移动遗传元件(MGEs),强调了它们在传播抗生素耐药性方面的重要性。基因组方法显示了毒力标记的多样化分布,包括用作水平基因转移工具的质粒和噬菌体序列。研究结果描述了在立陶宛多药耐药分离株中结合表型AMR检测对AMR和毒力遗传学决定因素进行的计算机预测。