Fattizzo Bruno, Serpenti Fabio, Barcellini Wilma, Caprioli Chiara
Hematology Unit. Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy.
Department of Oncology and Onco-Hematology, University of Milan, 20122 Milan, Italy.
Cancers (Basel). 2021 Jan 3;13(1):132. doi: 10.3390/cancers13010132.
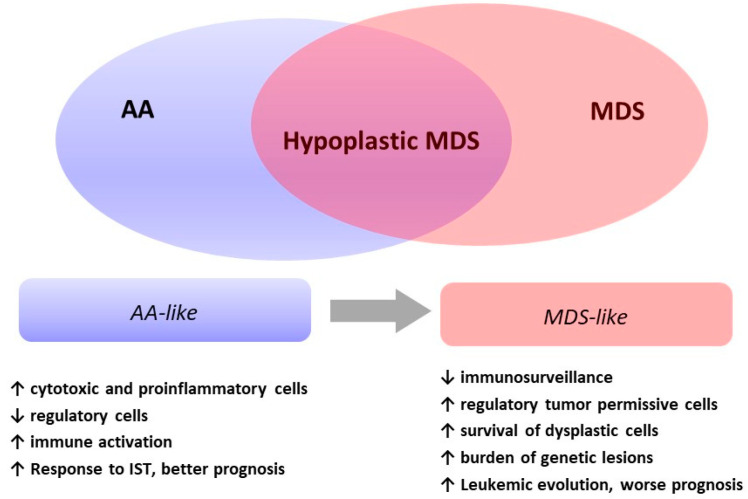
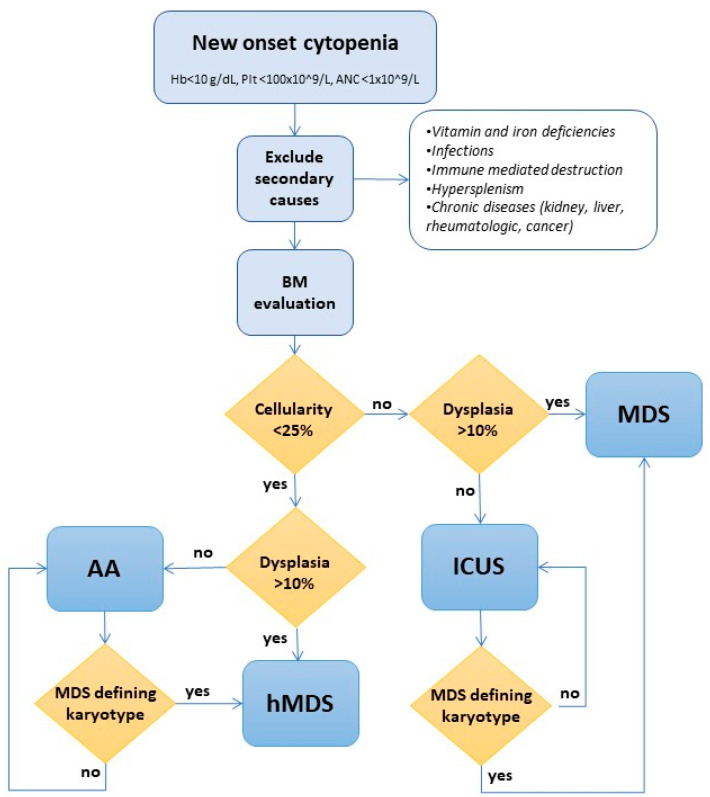
Myelodysplasias with hypocellular bone marrow (hMDS) represent about 10-15% of MDS and are defined by reduced bone marrow cellularity (i.e., <25% or an inappropriately reduced cellularity for their age in young patients). Their diagnosis is still an object of debate and has not been clearly established in the recent WHO classification. Clinical and morphological overlaps with both normo/hypercellular MDS and aplastic anemia include cytopenias, the presence of marrow hypocellularity and dysplasia, and cytogenetic and molecular alterations. Activation of the immune system against the hematopoietic precursors, typical of aplastic anemia, is reckoned even in hMDS and may account for the response to immunosuppressive treatment. Finally, the hMDS outcome seems more favorable than that of normo/hypercellular MDS patients. In this review, we analyze the available literature on hMDS, focusing on clinical, immunological, and molecular features. We show that hMDS pathogenesis and clinical presentation are peculiar, albeit in-between aplastic anemia (AA) and normo/hypercellular MDS. Two different hMDS phenotypes may be encountered: one featured by inflammation and immune activation, with increased cytotoxic T cells, increased T and B regulatory cells, and better response to immunosuppression; and the other, resembling MDS, where T and B regulatory/suppressor cells prevail, leading to genetic clonal selection and an increased risk of leukemic evolution. The identification of the prevailing hMDS phenotype might assist treatment choice, inform prognosis, and suggest personalized monitoring.
骨髓细胞减少的骨髓增生异常综合征(hMDS)约占骨髓增生异常综合征(MDS)的10%-15%,其定义为骨髓细胞减少(即<25%,或年轻患者中与其年龄不相称的细胞减少)。其诊断仍存在争议,在最近的世界卫生组织(WHO)分类中尚未明确确立。与正常细胞/高细胞MDS和再生障碍性贫血在临床和形态学上的重叠包括血细胞减少、骨髓细胞减少和发育异常的存在,以及细胞遗传学和分子改变。即使在hMDS中也认为存在针对造血前体细胞的免疫系统激活,这是再生障碍性贫血的典型特征,可能是对免疫抑制治疗有反应的原因。最后,hMDS的预后似乎比正常细胞/高细胞MDS患者更有利。在本综述中,我们分析了关于hMDS的现有文献,重点关注临床、免疫学和分子特征。我们表明,hMDS的发病机制和临床表现是独特的,尽管介于再生障碍性贫血(AA)和正常细胞/高细胞MDS之间。可能会遇到两种不同的hMDS表型:一种以炎症和免疫激活为特征,细胞毒性T细胞增加、T和B调节细胞增加,对免疫抑制的反应更好;另一种类似于MDS,其中T和B调节/抑制细胞占主导,导致基因克隆选择和白血病进展风险增加。确定占主导的hMDS表型可能有助于治疗选择、提供预后信息并建议进行个性化监测。