Pane Katia, Affinito Ornella, Zanfardino Mario, Castaldo Rossana, Incoronato Mariarosaria, Salvatore Marco, Franzese Monica
IRCCS SDN, Naples, Italy.
Front Genet. 2020 Dec 3;11:612521. doi: 10.3389/fgene.2020.612521. eCollection 2020.
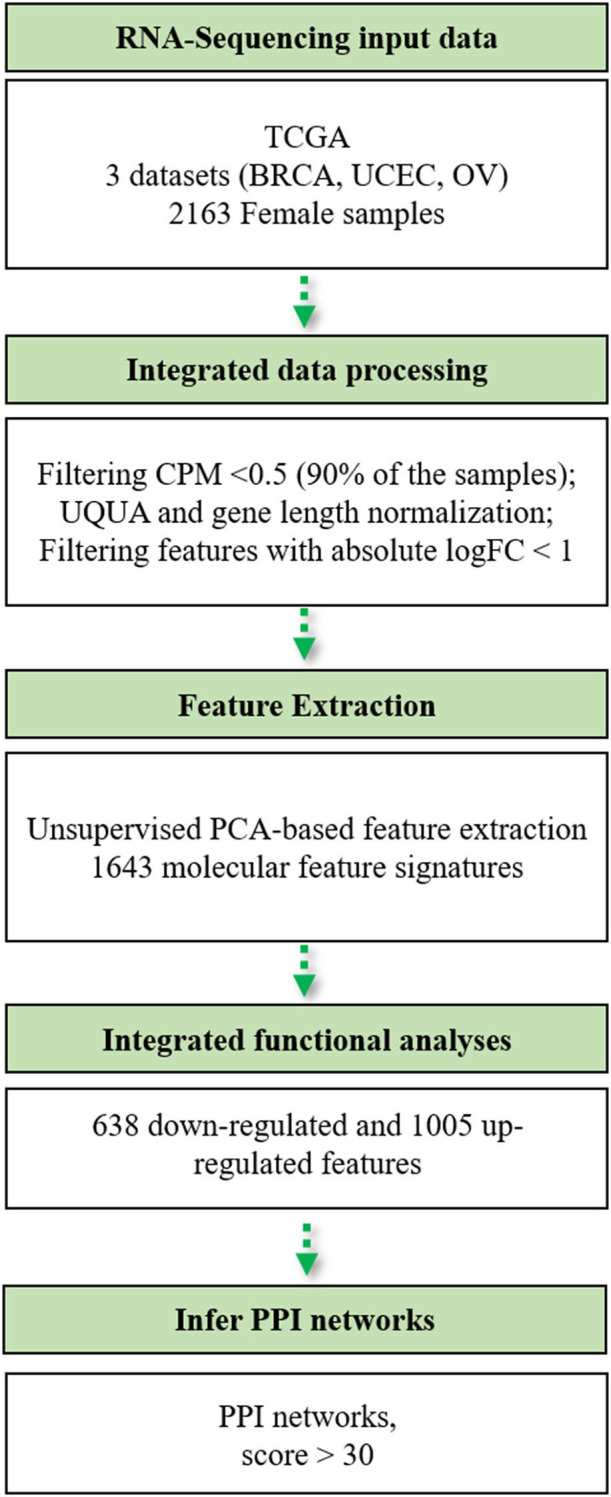
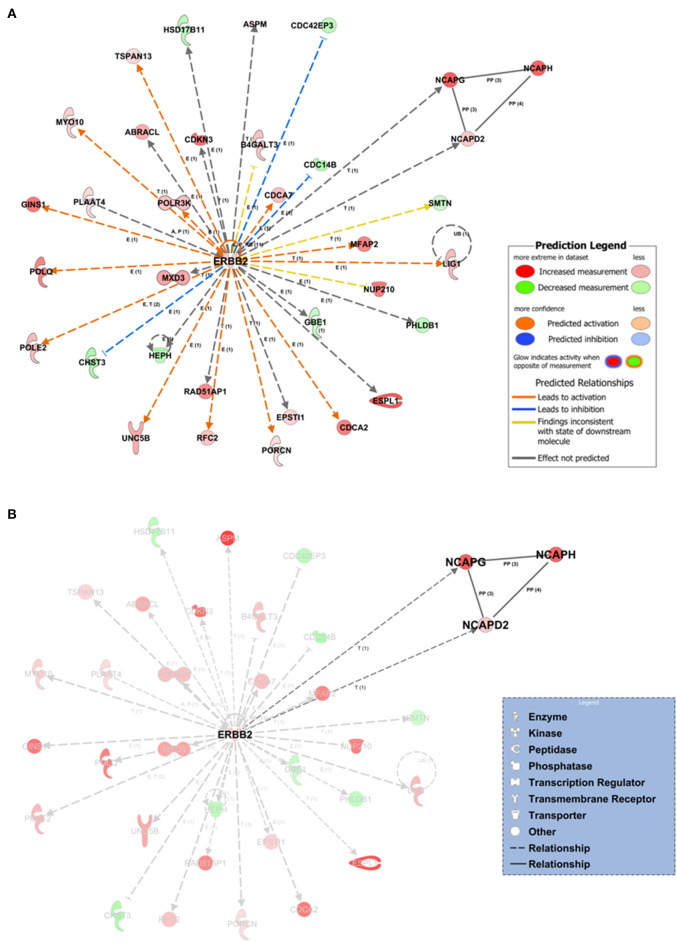
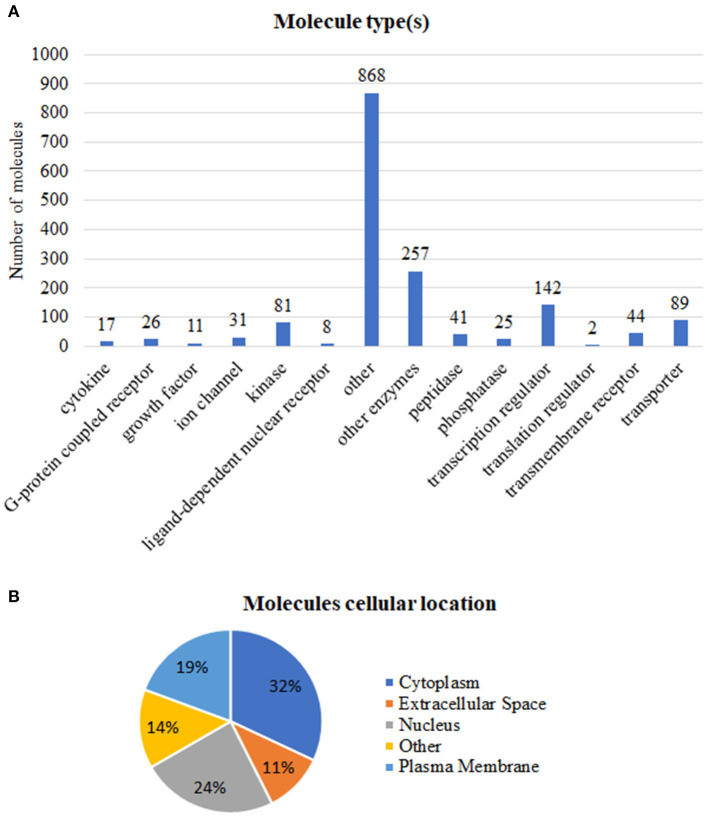
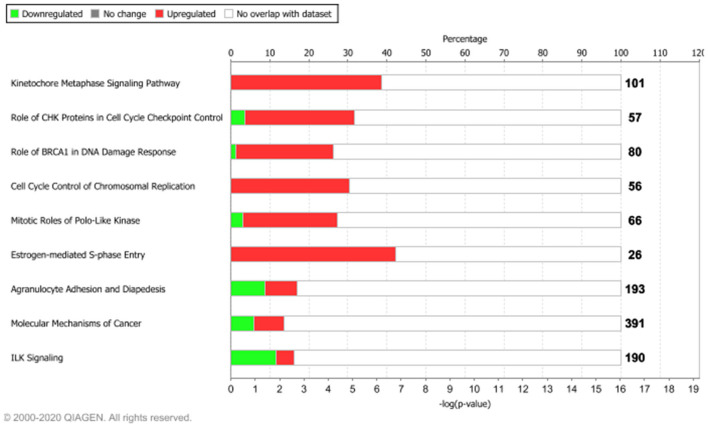
Breast, ovarian, and endometrial cancers have a major impact on mortality in women. These tumors share hormone-dependent mechanisms involved in female-specific cancers which support tumor growth in a different manner. Integrated computational approaches may allow us to better detect genomic similarities between these different female-specific cancers, helping us to deliver more sophisticated diagnosis and precise treatments. Recently, several initiatives of The Cancer Genome Atlas (TCGA) have encouraged integrated analyses of multiple cancers rather than individual tumors. These studies revealed common genetic alterations (driver genes) even in clinically distinct entities such as breast, ovarian, and endometrial cancers. In this study, we aimed to identify expression similarity signatures by extracting common genes among TCGA breast (BRCA), ovarian (OV), and uterine corpus endometrial carcinoma (UCEC) cohorts and infer co-regulatory protein-protein interaction networks that might have a relationship with the estrogen signaling pathway. Thus, we carried out an unsupervised principal component analysis (PCA)-based computational approach, using RNA sequencing data of 2,015 female cancer and 148 normal samples, in order to simultaneously capture the data heterogeneity of intertumors. Firstly, we identified tumor-associated genes from gene expression profiles. Secondly, we investigated the signaling pathways and co-regulatory protein-protein interaction networks underlying these three cancers by leveraging the Ingenuity Pathway Analysis software. In detail, we discovered 1,643 expression similarity signatures (638 downregulated and 1,005 upregulated genes, with respect to normal phenotype), denoted as tumor-associated genes. Through functional genomic analyses, we assessed that these genes were involved in the regulation of cell-cycle-dependent mechanisms, including metaphase kinetochore formation and estrogen-dependent S-phase entry. Furthermore, we generated putative co-regulatory protein-protein interaction networks, based on upstream regulators such as the ERBB2/HER2 gene. Moreover, we provided an bioinformatics workflow with a manageable list of intertumor expression similarity signatures for the three female-specific cancers. The expression similarity signatures identified in this study might uncover potential estrogen-dependent molecular mechanisms promoting carcinogenesis.
乳腺癌、卵巢癌和子宫内膜癌对女性死亡率有重大影响。这些肿瘤具有涉及女性特异性癌症的激素依赖性机制,这些机制以不同方式支持肿瘤生长。综合计算方法可能使我们能够更好地检测这些不同女性特异性癌症之间的基因组相似性,帮助我们进行更精确的诊断和精准治疗。最近,癌症基因组图谱(TCGA)的多项举措鼓励对多种癌症而非单个肿瘤进行综合分析。这些研究揭示了即使在临床特征不同的实体(如乳腺癌、卵巢癌和子宫内膜癌)中也存在共同的基因改变(驱动基因)。在本研究中,我们旨在通过提取TCGA乳腺癌(BRCA)、卵巢癌(OV)和子宫内膜癌(UCEC)队列中的共同基因来识别表达相似性特征,并推断可能与雌激素信号通路相关的共调控蛋白质-蛋白质相互作用网络。因此,我们使用2015例女性癌症和148例正常样本的RNA测序数据,开展了一种基于无监督主成分分析(PCA)的计算方法,以便同时捕捉肿瘤间的数据异质性。首先,我们从基因表达谱中识别出肿瘤相关基因。其次,我们利用Ingenuity Pathway Analysis软件研究这三种癌症潜在的信号通路和共调控蛋白质-蛋白质相互作用网络。具体而言,我们发现了1643个表达相似性特征(相对于正常表型,638个下调基因和1005个上调基因),将其称为肿瘤相关基因。通过功能基因组分析,我们评估这些基因参与了细胞周期依赖性机制的调控,包括中期动粒形成和雌激素依赖性S期进入。此外,我们基于ERBB2/HER2基因等上游调节因子生成了假定的共调控蛋白质-蛋白质相互作用网络。而且,我们为这三种女性特异性癌症提供了一个具有可管理的肿瘤间表达相似性特征列表的生物信息学工作流程。本研究中识别出的表达相似性特征可能揭示促进致癌作用的潜在雌激素依赖性分子机制。