Department of Obstetrics and Gynecology, Tohoku University Graduate School of Medicine, Sendai, Japan.
Advanced Research Center for Innovations in Next-Generation Medicine, Tohoku University, Sendai, Japan.
PLoS One. 2021 Jan 11;16(1):e0236907. doi: 10.1371/journal.pone.0236907. eCollection 2021.
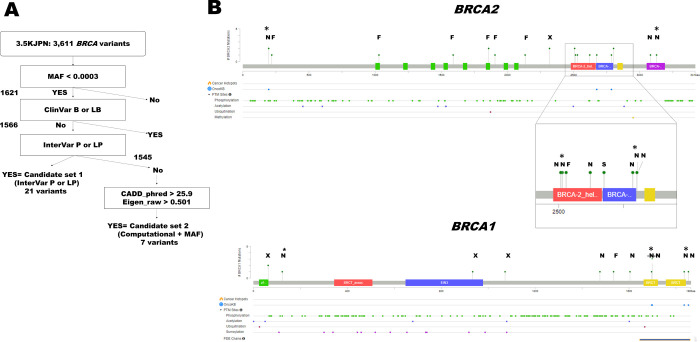
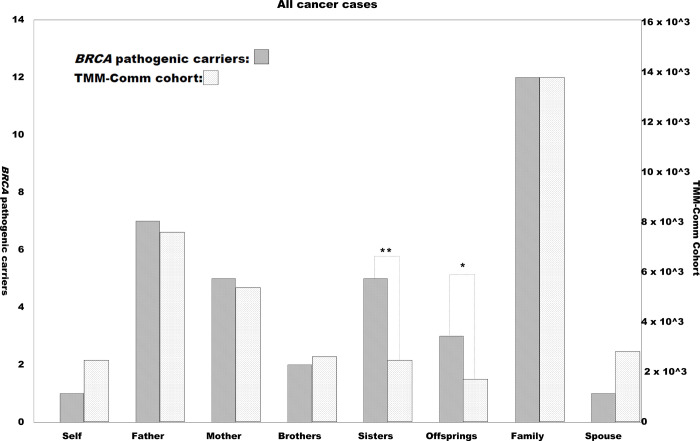
Identification of the population frequencies of definitely pathogenic germline variants in two major hereditary breast and ovarian cancer syndrome (HBOC) genes, BRCA1/2, is essential to estimate the number of HBOC patients. In addition, the identification of moderately penetrant HBOC gene variants that contribute to increasing the risk of breast and ovarian cancers in a population is critical to establish personalized health care. A prospective cohort subjected to genome analysis can provide both sets of information. Computational scoring and prospective cohort studies may help to identify such likely pathogenic variants in the general population. We annotated the variants in the BRCA1 and BRCA2 genes from a dataset of 3,552 whole-genome sequences obtained from members of a prospective cohorts with genome data in the Tohoku Medical Megabank Project (TMM) with InterVar software. Computational impact scores (CADD_phred and Eigen_raw) and minor allele frequencies (MAFs) of pathogenic (P) and likely pathogenic (LP) variants in ClinVar were used for filtration criteria. Familial predispositions to cancers among the 35,000 TMM genome cohort participants were analyzed to verify the identified pathogenicity. Seven potentially pathogenic variants were newly identified. The sisters of carriers of these moderately deleterious variants and definite P and LP variants among members of the TMM prospective cohort showed a statistically significant preponderance for cancer onset, from the self-reported cancer history. Filtering by computational scoring and MAF is useful to identify potentially pathogenic variants in BRCA genes in the Japanese population. These results should help to follow up the carriers of variants of uncertain significance in the HBOC genes in the longitudinal prospective cohort study.
确定两个主要遗传性乳腺癌和卵巢癌综合征 (HBOC) 基因 BRCA1/2 中的种系致病性变异的人群频率对于估计 HBOC 患者的数量至关重要。此外,确定中度外显率的 HBOC 基因变异,这些变异会增加人群中乳腺癌和卵巢癌的风险,对于建立个性化医疗保健至关重要。接受基因组分析的前瞻性队列可以提供这两组信息。计算评分和前瞻性队列研究可能有助于在普通人群中识别此类可能的致病性变异。我们使用 InterVar 软件对来自东北医疗巨型银行项目 (TMM) 基因组数据的前瞻性队列成员的 3552 个全基因组序列数据集的 BRCA1 和 BRCA2 基因中的变体进行注释。计算影响评分 (CADD_phred 和 Eigen_raw) 和 ClinVar 中致病性 (P) 和可能致病性 (LP) 变体的次要等位基因频率 (MAFs) 用于过滤标准。分析了 35000 名 TMM 基因组队列参与者中的癌症家族易感性,以验证鉴定的致病性。新确定了七个潜在致病性变体。这些中度有害变体携带者的姐妹,以及 TMM 前瞻性队列成员中确定的 P 和 LP 变体携带者的姐妹,从自我报告的癌症史来看,癌症发病的倾向具有统计学意义。通过计算评分和 MAF 过滤有助于识别日本人群中 BRCA 基因中的潜在致病性变体。这些结果应有助于在纵向前瞻性队列研究中对 HBOC 基因中不确定意义的变体携带者进行随访。