ESAT-STADIUS, KU Leuven, Leuven, 3001, Belgium.
Università di Torino, Torino, Italy, Torino, 10123, Italy.
BMC Biol. 2021 Jan 13;19(1):3. doi: 10.1186/s12915-020-00930-0.
Identifying variants that drive tumor progression (driver variants) and distinguishing these from variants that are a byproduct of the uncontrolled cell growth in cancer (passenger variants) is a crucial step for understanding tumorigenesis and precision oncology. Various bioinformatics methods have attempted to solve this complex task.
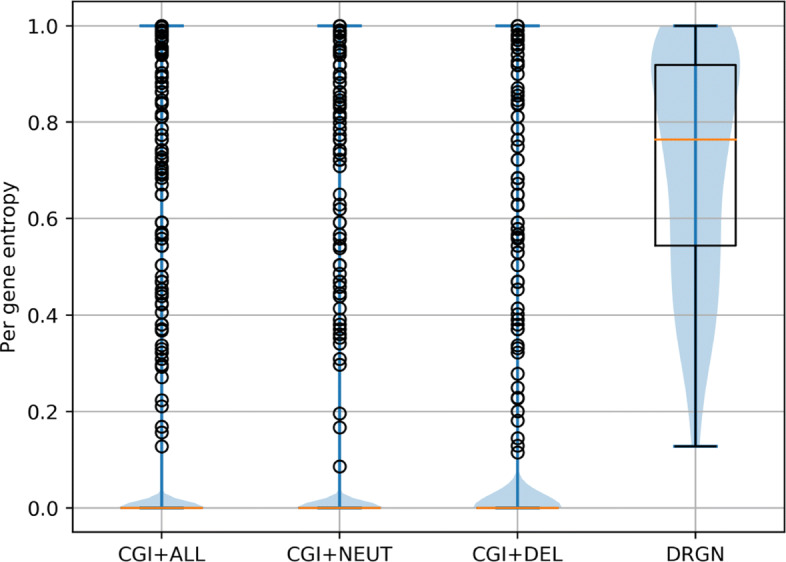
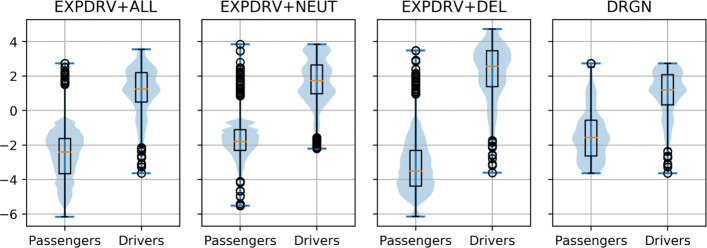
In this study, we investigate the assumptions on which these methods are based, showing that the different definitions of driver and passenger variants influence the difficulty of the prediction task. More importantly, we prove that the data sets have a construction bias which prevents the machine learning (ML) methods to actually learn variant-level functional effects, despite their excellent performance. This effect results from the fact that in these data sets, the driver variants map to a few driver genes, while the passenger variants spread across thousands of genes, and thus just learning to recognize driver genes provides almost perfect predictions.
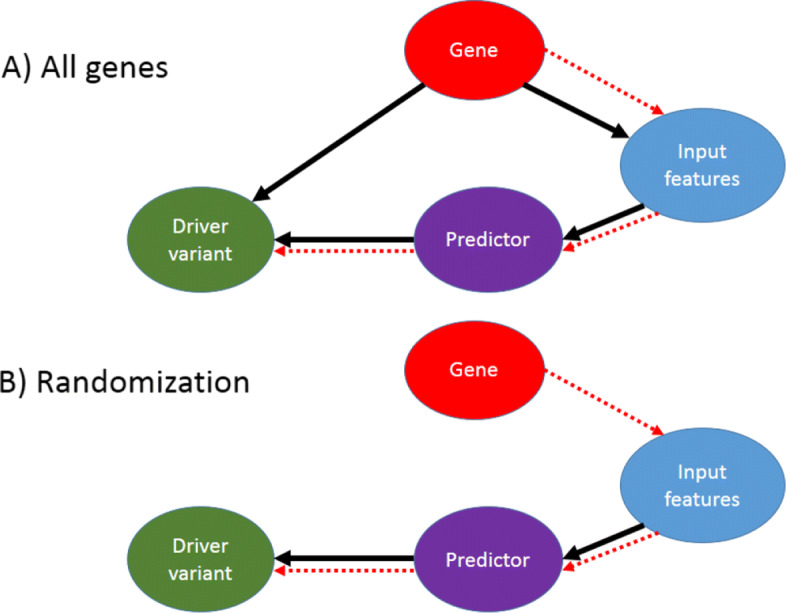
To mitigate this issue, we propose a novel data set that minimizes this bias by ensuring that all genes covered by the data contain both driver and passenger variants. As a result, we show that the tested predictors experience a significant drop in performance, which should not be considered as poorer modeling, but rather as correcting unwarranted optimism. Finally, we propose a weighting procedure to completely eliminate the gene effects on such predictions, thus precisely evaluating the ability of predictors to model the functional effects of single variants, and we show that indeed this task is still open.
识别驱动肿瘤进展的变异(驱动变异),并将其与癌症失控细胞生长的副产品变异(乘客变异)区分开来,是理解肿瘤发生和精准肿瘤学的关键步骤。各种生物信息学方法都试图解决这一复杂任务。
在这项研究中,我们研究了这些方法所基于的假设,表明驱动变异和乘客变异的不同定义会影响预测任务的难度。更重要的是,我们证明了数据集存在构建偏差,这使得机器学习(ML)方法无法真正学习变异级别的功能效应,尽管它们的性能非常出色。这种效应是由于在这些数据集中,驱动变异映射到少数几个驱动基因,而乘客变异则分布在数千个基因中,因此仅仅学习识别驱动基因就可以提供几乎完美的预测。
为了解决这个问题,我们提出了一个新的数据集,通过确保数据涵盖的所有基因都包含驱动变异和乘客变异,来最小化这种偏差。结果表明,测试的预测器的性能显著下降,这不应该被视为较差的建模,而应该被视为纠正不必要的乐观。最后,我们提出了一种加权程序,可以完全消除基因对这些预测的影响,从而精确评估预测器对单个变异的功能效应进行建模的能力,我们确实表明,这项任务仍然存在。