Pagel Kymberleigh A, Kim Rick, Moad Kyle, Busby Ben, Zheng Lily, Tokheim Collin, Ryan Michael, Karchin Rachel
The Institute for Computational Medicine, The Johns Hopkins University, Baltimore, MD.
In Silico Solutions, Falls Church, VA.
JCO Clin Cancer Inform. 2020 Mar;4:310-317. doi: 10.1200/CCI.19.00132.
The modern researcher is confronted with hundreds of published methods to interpret genetic variants. There are databases of genes and variants, phenotype-genotype relationships, algorithms that score and rank genes, and in silico variant effect prediction tools. Because variant prioritization is a multifactorial problem, a welcome development in the field has been the emergence of decision support frameworks, which make it easier to integrate multiple resources in an interactive environment. Current decision support frameworks are typically limited by closed proprietary architectures, access to a restricted set of tools, lack of customizability, Web dependencies that expose protected data, or limited scalability.
We present the Open Custom Ranked Analysis of Variants Toolkit (OpenCRAVAT) a new open-source, scalable decision support system for variant and gene prioritization. We have designed the resource catalog to be open and modular to maximize community and developer involvement, and as a result, the catalog is being actively developed and growing every month. Resources made available via the store are well suited for analysis of cancer, as well as Mendelian and complex diseases.
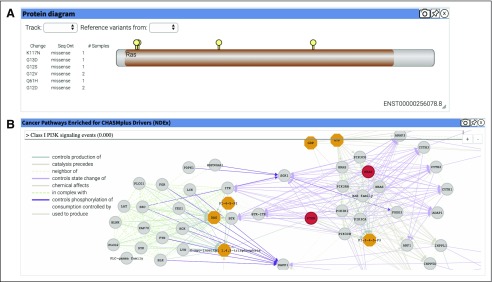
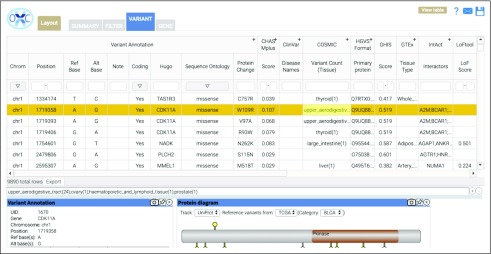
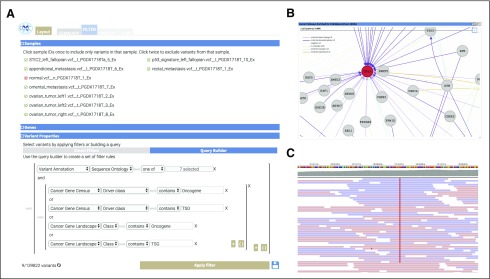
OpenCRAVAT offers both command-line utility and dynamic graphical user interface, allowing users to install with a single command, easily download tools from an extensive resource catalog, create customized pipelines, and explore results in a richly detailed viewing environment. We present several case studies to illustrate the design of custom workflows to prioritize genes and variants.
OpenCRAVAT is distinguished from similar tools by its capabilities to access and integrate an unprecedented amount of diverse data resources and computational prediction methods, which span germline, somatic, common, rare, coding, and noncoding variants.
现代研究人员面临着数百种已发表的解释基因变异的方法。有基因和变异数据库、表型-基因型关系、对基因进行评分和排序的算法以及计算机模拟变异效应预测工具。由于变异优先级排序是一个多因素问题,该领域一个受欢迎的发展是决策支持框架的出现,它使得在交互式环境中整合多种资源变得更加容易。当前的决策支持框架通常受到封闭的专有架构、对一组受限工具的访问、缺乏可定制性、暴露受保护数据的网络依赖性或有限的可扩展性的限制。
我们展示了变异体和基因优先级排序的开放定制排序分析工具包(OpenCRAVAT),这是一个新的开源、可扩展的决策支持系统。我们将资源目录设计为开放且模块化的,以最大限度地提高社区和开发者的参与度,因此,该目录正在积极开发且每月都在增长。通过该存储库提供资源非常适合癌症以及孟德尔和复杂疾病的分析。
OpenCRAVAT提供命令行实用程序和动态图形用户界面,允许用户通过单个命令进行安装,轻松从广泛的资源目录中下载工具,创建定制管道,并在丰富详细的查看环境中探索结果。我们展示了几个案例研究,以说明用于对基因和变异进行优先级排序的定制工作流程的设计。
OpenCRAVAT与类似工具的不同之处在于,它能够访问和整合前所未有的大量多样的数据资源和计算预测方法,这些资源和方法涵盖种系、体细胞、常见、罕见、编码和非编码变异。