Division of Medical Genetics, Department of Medicine, University of California San Diego, La Jolla, CA, USA.
Bioinformatics and Systems Biology Program, University of California San Diego, La Jolla, CA, USA.
Hum Genet. 2022 Jun;141(6):1195-1210. doi: 10.1007/s00439-021-02329-5. Epub 2021 Aug 25.
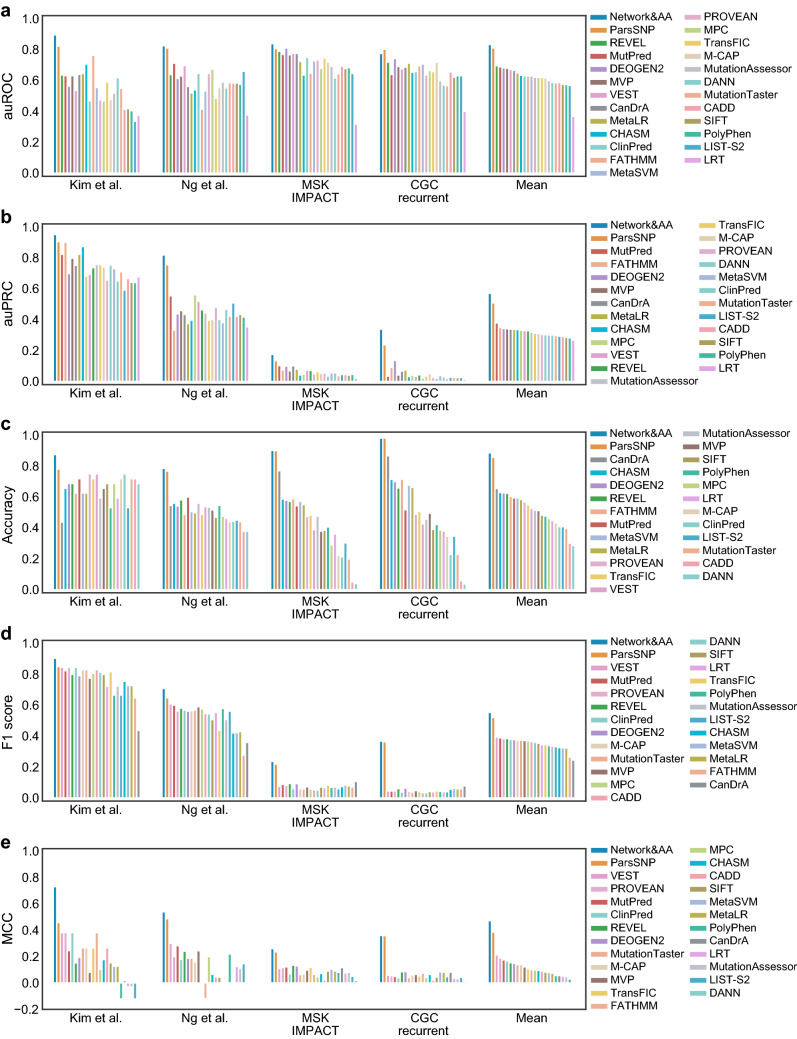
Variant interpretation remains a central challenge for precision medicine. Missense variants are particularly difficult to understand as they change only a single amino acid in a protein sequence yet can have large and varied effects on protein activity. Numerous tools have been developed to identify missense variants with putative disease consequences from protein sequence and structure. However, biological function arises through higher order interactions among proteins and molecules within cells. We therefore sought to capture information about the potential of missense mutations to perturb protein interaction networks by integrating protein structure and interaction data. We developed 16 network-based annotations for missense mutations that provide orthogonal information to features classically used to prioritize variants. We then evaluated them in the context of a proven machine-learning framework for variant effect prediction across multiple benchmark datasets to demonstrate their potential to improve variant classification. Interestingly, network features resulted in larger performance gains for classifying somatic mutations than for germline variants, possibly due to different constraints on what mutations are tolerated at the cellular versus organismal level. Our results suggest that modeling variant potential to perturb context-specific interactome networks is a fruitful strategy to advance in silico variant effect prediction.
变异解释仍然是精准医学的一个核心挑战。错义变异特别难以理解,因为它们只在蛋白质序列中改变一个氨基酸,但对蛋白质活性的影响可能很大且多样。已经开发了许多工具来从蛋白质序列和结构中识别具有潜在疾病后果的错义变异。然而,生物功能是通过细胞内蛋白质和分子之间的更高阶相互作用产生的。因此,我们试图通过整合蛋白质结构和相互作用数据来捕捉错义突变干扰蛋白质相互作用网络的潜力。我们开发了 16 种基于网络的错义突变注释,为经典用于优先排序变异的特征提供了正交信息。然后,我们在经过验证的机器学习框架中评估了它们在多个基准数据集上的变体效应预测,以证明它们在改善变体分类方面的潜力。有趣的是,网络特征在分类体细胞突变方面比在分类种系变体方面产生了更大的性能提升,这可能是由于在细胞和生物体水平上对突变的容忍度不同。我们的结果表明,模拟变异干扰特定于上下文的互作网络的潜力是推进计算机变异效应预测的一种富有成效的策略。