Department of Computer Science, Brown University, Providence, RI, USA.
Department of Computer Science, Princeton University, Princeton, NJ, USA.
Bioinformatics. 2017 Jul 15;33(14):i152-i160. doi: 10.1093/bioinformatics/btx270.
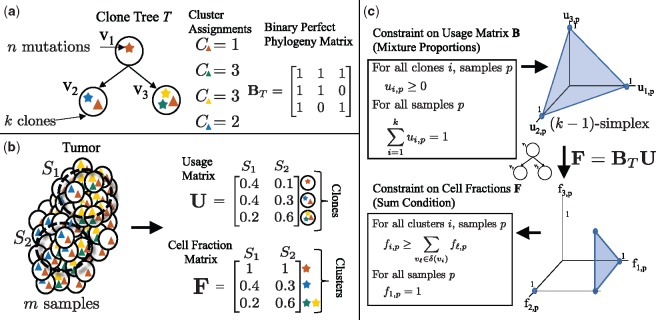
A tumor arises from an evolutionary process that can be modeled as a phylogenetic tree. However, reconstructing this tree is challenging as most cancer sequencing uses bulk tumor tissue containing heterogeneous mixtures of cells.
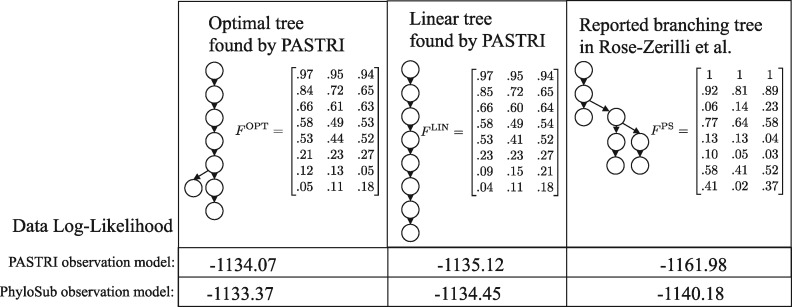
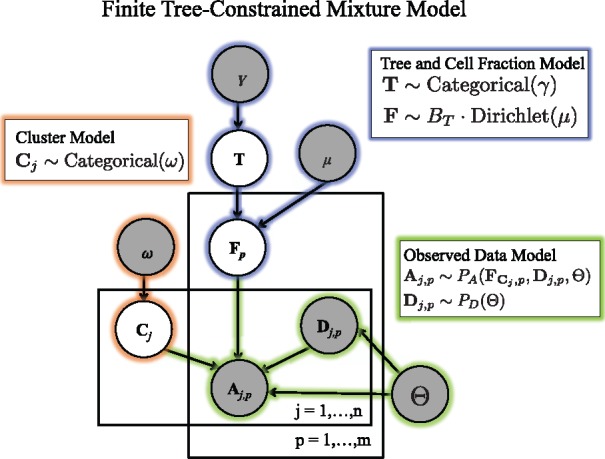
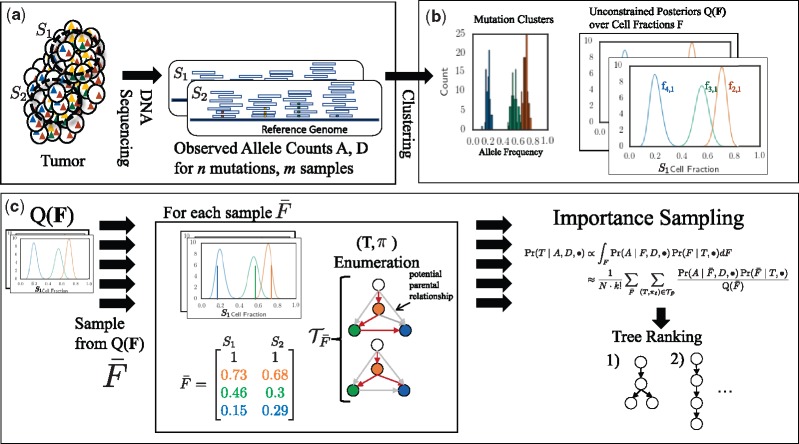
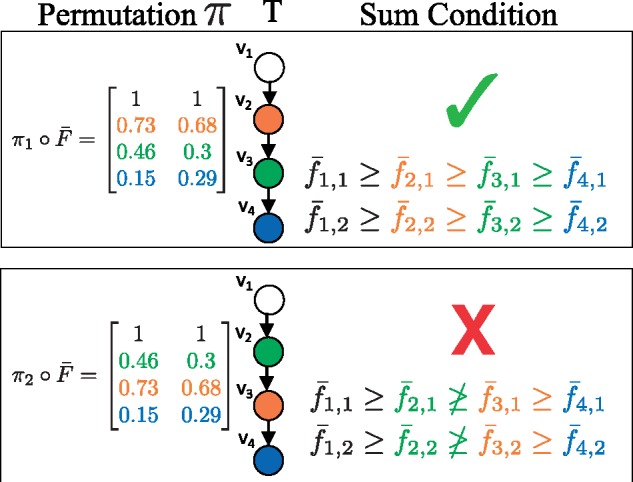
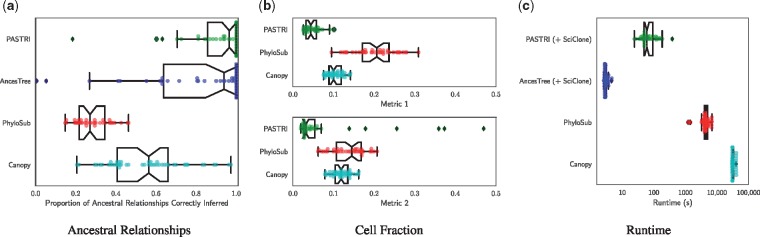
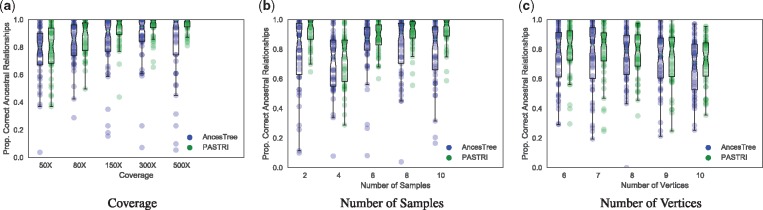
We introduce P robabilistic A lgorithm for S omatic Tr ee I nference (PASTRI), a new algorithm for bulk-tumor sequencing data that clusters somatic mutations into clones and infers a phylogenetic tree that describes the evolutionary history of the tumor. PASTRI uses an importance sampling algorithm that combines a probabilistic model of DNA sequencing data with a enumeration algorithm based on the combinatorial constraints defined by the underlying phylogenetic tree. As a result, tree inference is fast, accurate and robust to noise. We demonstrate on simulated data that PASTRI outperforms other cancer phylogeny algorithms in terms of runtime and accuracy. On real data from a chronic lymphocytic leukemia (CLL) patient, we show that a simple linear phylogeny better explains the data the complex branching phylogeny that was previously reported. PASTRI provides a robust approach for phylogenetic tree inference from mixed samples.
Software is available at compbio.cs.brown.edu/software.
Supplementary data are available at Bioinformatics online.
肿瘤起源于进化过程,可以将其建模为系统发育树。然而,由于大多数癌症测序使用包含异质细胞混合物的肿瘤组织进行批量测序,因此重建该树具有挑战性。
我们引入了用于批量肿瘤测序数据的概率算法,即体细胞树推断 (PASTRI),它将体细胞突变聚类到克隆中,并推断出描述肿瘤进化历史的系统发育树。PASTRI 使用一种重要性抽样算法,该算法将 DNA 测序数据的概率模型与基于基础系统发育树定义的组合约束的枚举算法相结合。因此,树推断速度快、准确且对噪声具有鲁棒性。我们在模拟数据上证明,PASTRI 在运行时间和准确性方面优于其他癌症系统发育算法。在来自慢性淋巴细胞白血病 (CLL) 患者的真实数据上,我们表明简单的线性系统发育更好地解释了数据,而之前报道的复杂分支系统发育则较差。PASTRI 为从混合样本中推断系统发育树提供了一种稳健的方法。
软件可在 compbio.cs.brown.edu/software 获得。
补充数据可在 Bioinformatics 在线获得。