Zhao Xingwang, Zhang Longlong, Wang Juan, Zhang Min, Song Zhiqiang, Ni Bing, You Yi
Department of Dermatology, Southwest Hospital, Army Medical University, (Third Military Medical University), Chongqing, 400038, China.
State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, 650223, China.
J Transl Med. 2021 Jan 19;19(1):35. doi: 10.1186/s12967-020-02698-x.
Systemic lupus erythematosus (SLE) is a multisystemic, chronic inflammatory disease characterized by destructive systemic organ involvement, which could cause the decreased functional capacity, increased morbidity and mortality. Previous studies show that SLE is characterized by autoimmune, inflammatory processes, and tissue destruction. Some seriously-ill patients could develop into lupus nephritis. However, the cause and underlying molecular events of SLE needs to be further resolved.
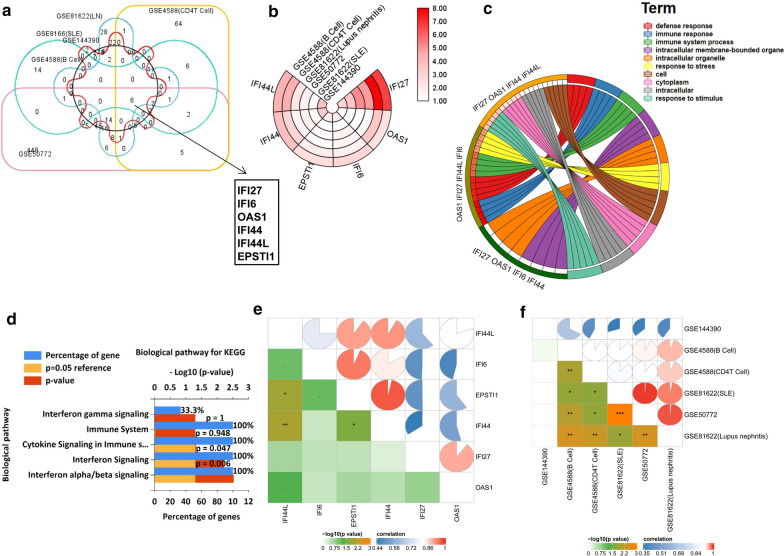
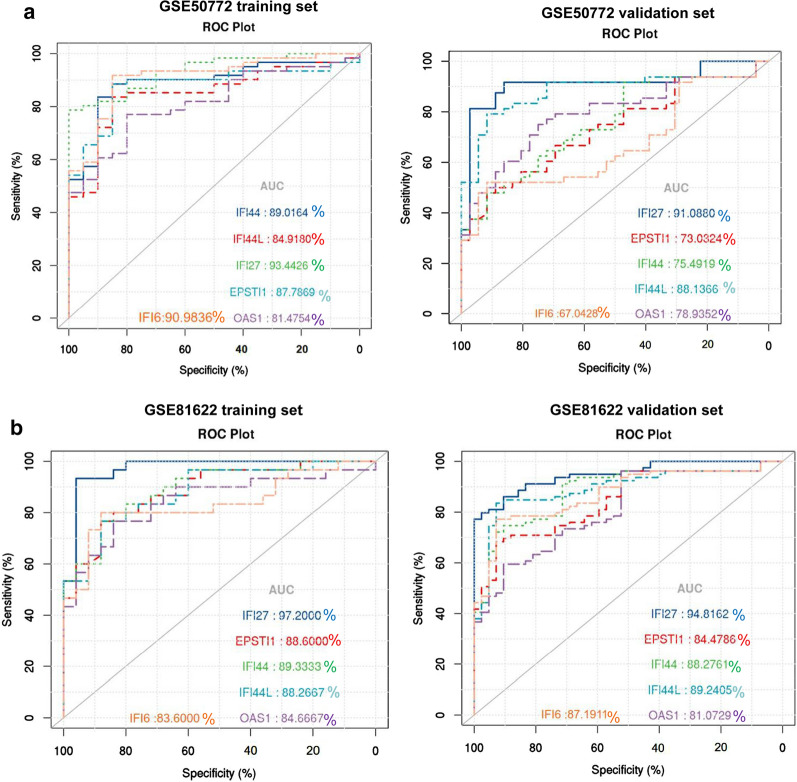
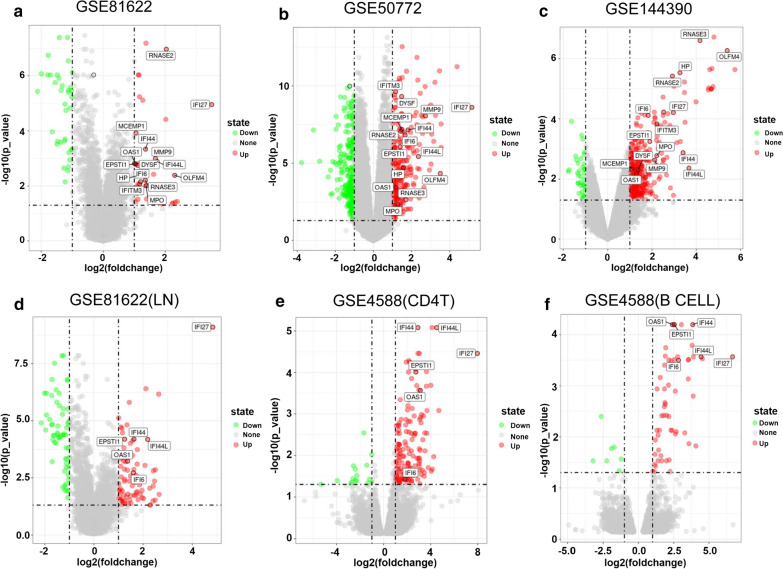
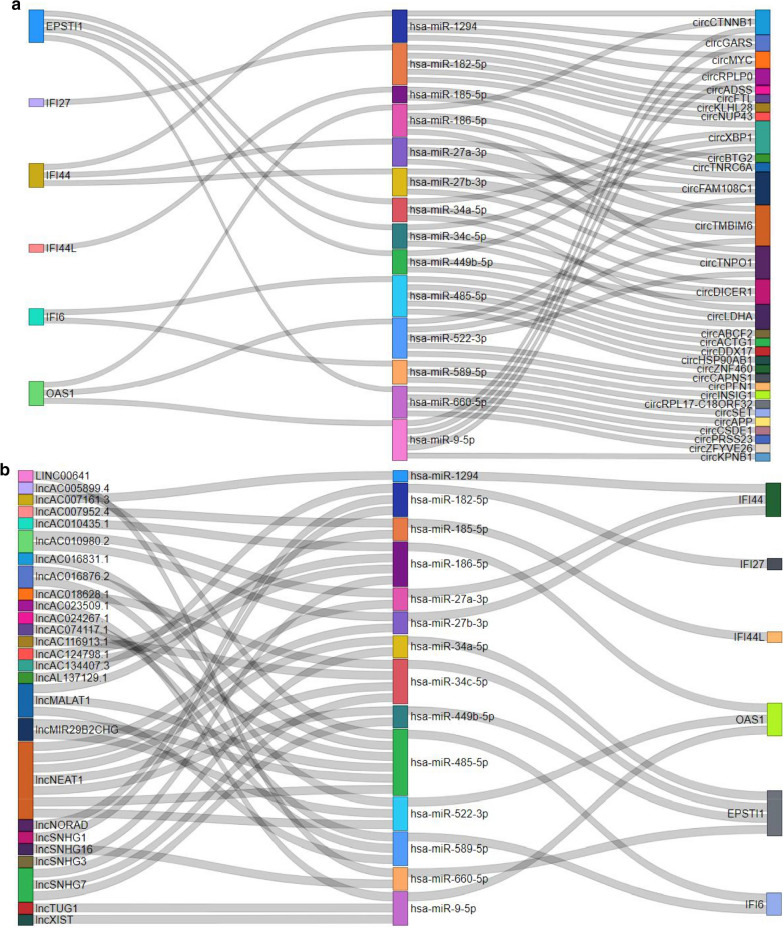
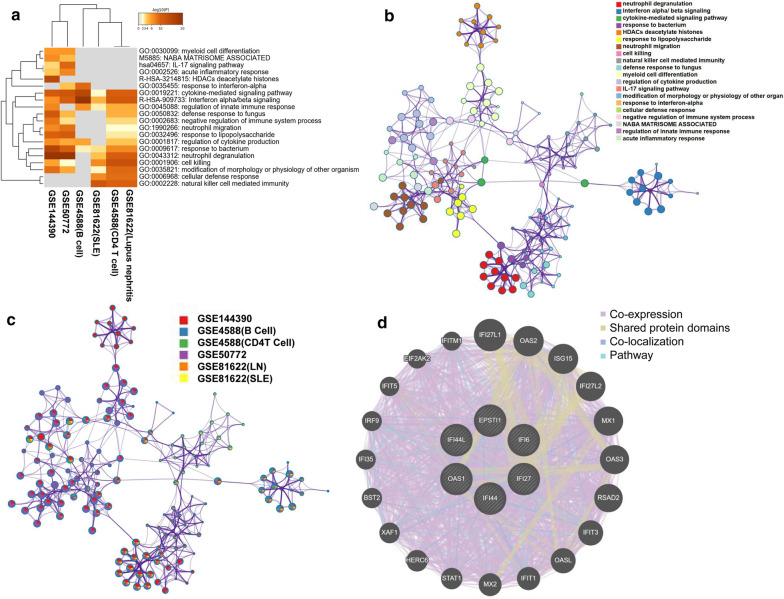
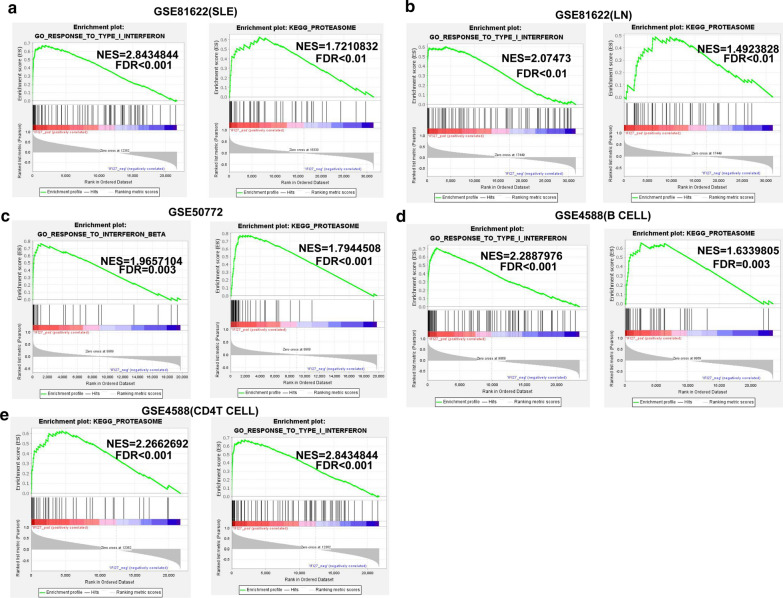
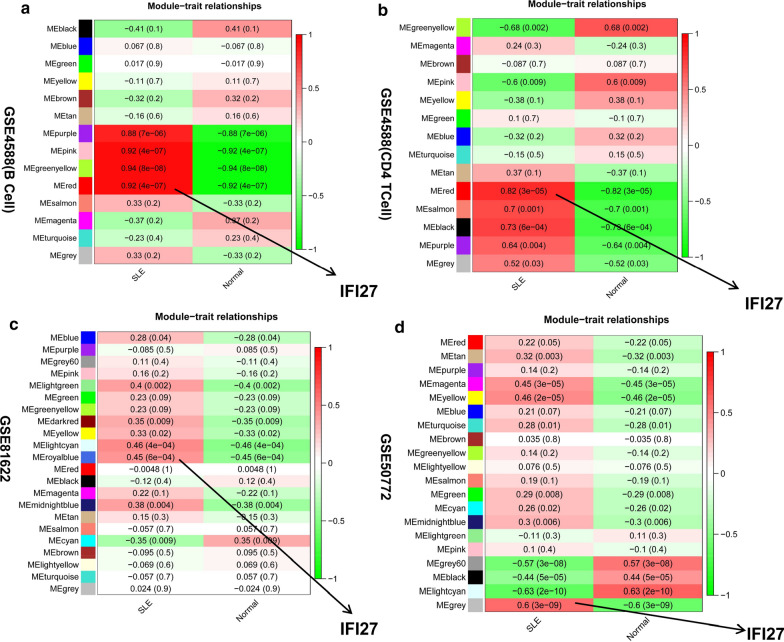
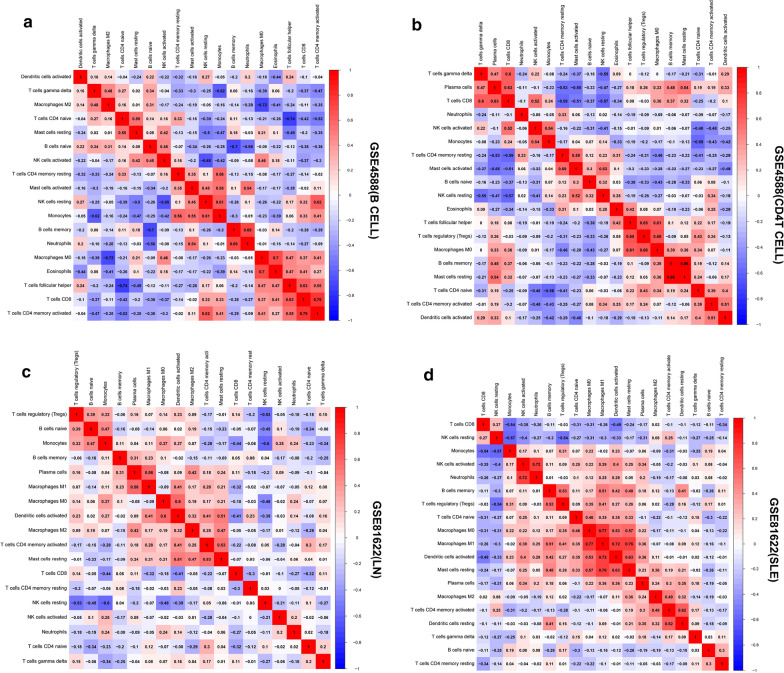
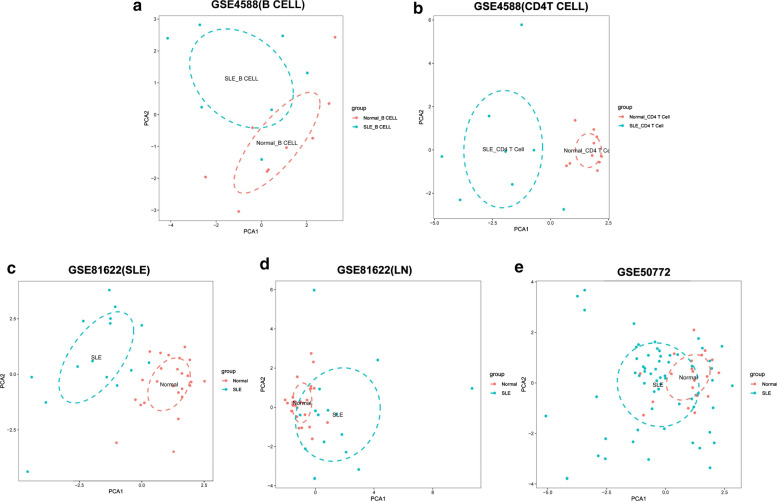
The expression profiles of GSE144390, GSE4588, GSE50772 and GSE81622 were downloaded from the Gene Expression Omnibus (GEO) database to obtain differentially expressed genes (DEGs) between SLE and healthy samples. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichments of DEGs were performed by metascape etc. online analyses. The protein-protein interaction (PPI) networks of the DEGs were constructed by GENEMANIA software. We performed Gene Set Enrichment Analysis (GSEA) to further understand the functions of the hub gene, Weighted gene co-expression network analysis (WGCNA) would be utilized to build a gene co-expression network, and the most significant module and hub genes was identified. CIBERSORT tools have facilitated the analysis of immune cell infiltration patterns of diseases. The receiver operating characteristic (ROC) analyses were conducted to explore the value of DEGs for SLE diagnosis.
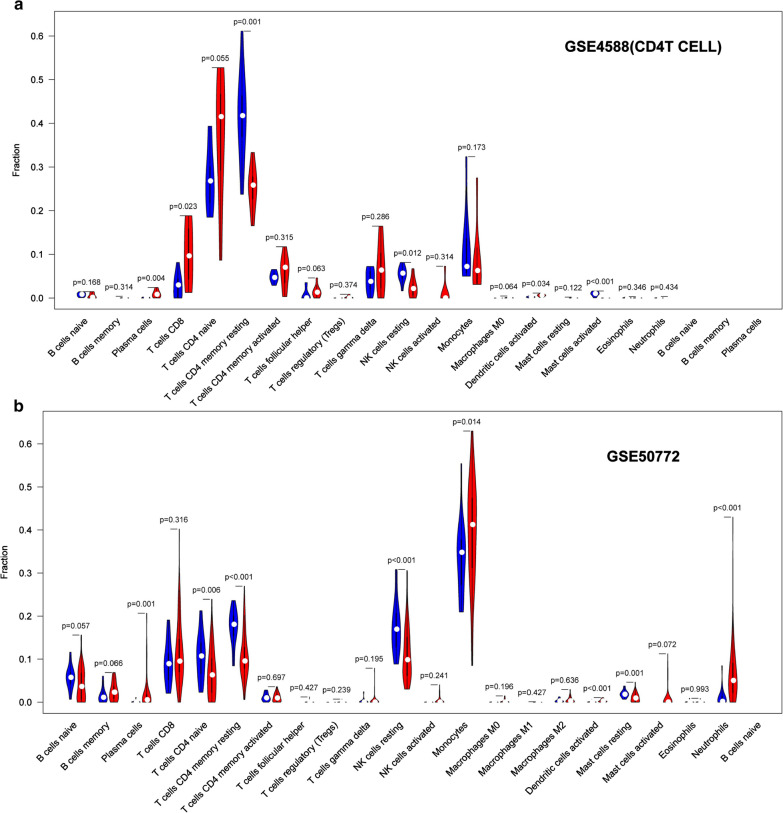
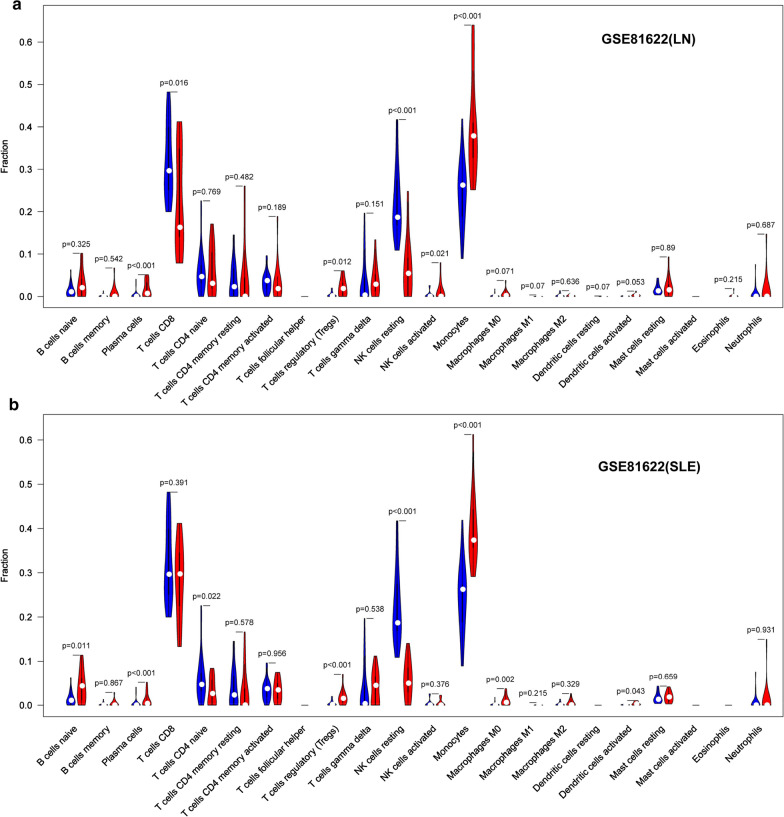
In total, 6 DEGs (IFI27, IFI44, IFI44L, IFI6, EPSTI1 and OAS1) were screened, Biological functions analysis identified key related pathways, gene modules and co-expression networks in SLE. IFI27 may be closely correlated with the occurrence of SLE. We found that an increased infiltration of moncytes, while NK cells resting infiltrated less may be related to the occurrence of SLE.
IFI27 may be closely related pathogenesis of SLE, and represents a new candidate molecular marker of the occurrence and progression of SLE. Moreover immune cell infiltration plays important role in the progession of SLE.
系统性红斑狼疮(SLE)是一种多系统慢性炎症性疾病,其特征为系统性器官受累并具有破坏性,可导致功能能力下降、发病率和死亡率增加。既往研究表明,SLE具有自身免疫、炎症过程和组织破坏的特征。一些重症患者可能会发展为狼疮性肾炎。然而,SLE的病因及潜在分子事件仍有待进一步明确。
从基因表达综合数据库(GEO)下载GSE144390、GSE4588、GSE50772和GSE81622的表达谱,以获取SLE与健康样本之间的差异表达基因(DEG)。通过metascape等在线分析对DEG进行基因本体(GO)和京都基因与基因组百科全书(KEGG)通路富集分析。利用GENEMANIA软件构建DEG的蛋白质-蛋白质相互作用(PPI)网络。我们进行基因集富集分析(GSEA)以进一步了解枢纽基因的功能,利用加权基因共表达网络分析(WGCNA)构建基因共表达网络,并确定最显著的模块和枢纽基因。CIBERSORT工具有助于分析疾病的免疫细胞浸润模式。进行受试者工作特征(ROC)分析以探索DEG对SLE诊断的价值。
共筛选出6个DEG(IFI27、IFI44、IFI44L、IFI6、EPSTI1和OAS1),生物学功能分析确定了SLE中的关键相关通路、基因模块和共表达网络。IFI27可能与SLE的发生密切相关。我们发现单核细胞浸润增加,而静息自然杀伤细胞浸润减少可能与SLE的发生有关。
IFI27可能与SLE的发病机制密切相关,是SLE发生和进展的新候选分子标志物。此外,免疫细胞浸润在SLE的进展中起重要作用。