Drug Metabolism and Pharmacokinetics, AbbVie Bioresearch Center, Worcester, MA, 01605, USA.
Discovery and Exploratory Statistics, AbbVie Bioresearch Center, Worcester, MA, 01605, USA.
Sci Rep. 2021 Jan 19;11(1):1760. doi: 10.1038/s41598-021-81279-4.
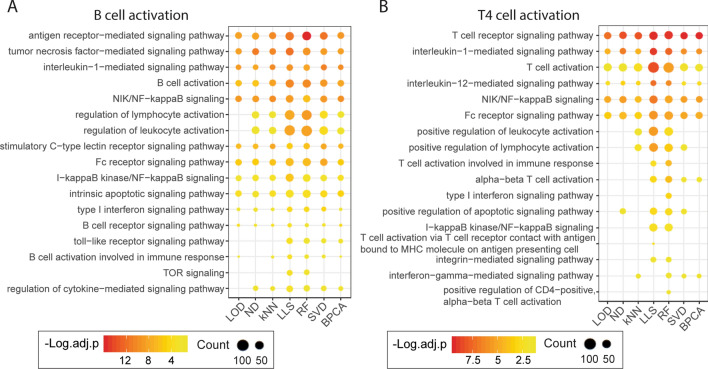
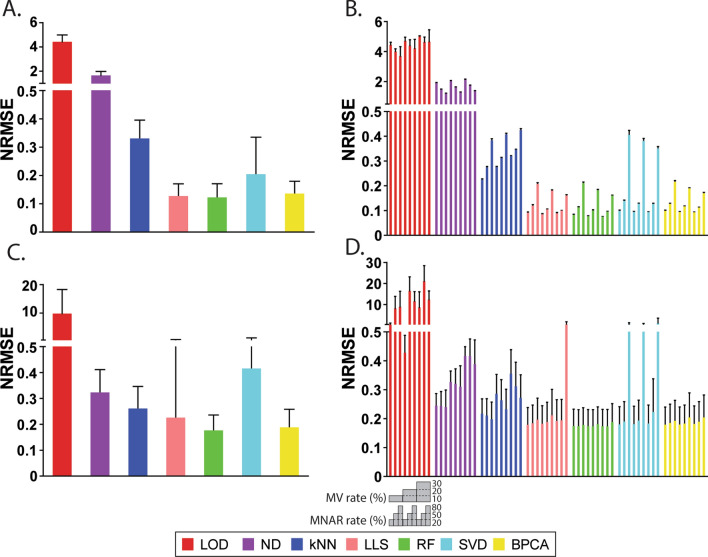
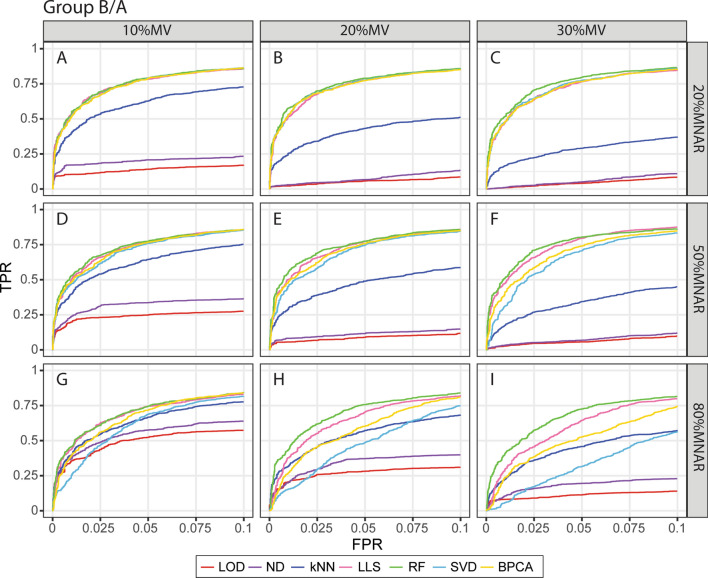
The presence of missing values (MVs) in label-free quantitative proteomics greatly reduces the completeness of data. Imputation has been widely utilized to handle MVs, and selection of the proper method is critical for the accuracy and reliability of imputation. Here we present a comparative study that evaluates the performance of seven popular imputation methods with a large-scale benchmark dataset and an immune cell dataset. Simulated MVs were incorporated into the complete part of each dataset with different combinations of MV rates and missing not at random (MNAR) rates. Normalized root mean square error (NRMSE) was applied to evaluate the accuracy of protein abundances and intergroup protein ratios after imputation. Detection of true positives (TPs) and false altered-protein discovery rate (FADR) between groups were also compared using the benchmark dataset. Furthermore, the accuracy of handling real MVs was assessed by comparing enriched pathways and signature genes of cell activation after imputing the immune cell dataset. We observed that the accuracy of imputation is primarily affected by the MNAR rate rather than the MV rate, and downstream analysis can be largely impacted by the selection of imputation methods. A random forest-based imputation method consistently outperformed other popular methods by achieving the lowest NRMSE, high amount of TPs with the average FADR < 5%, and the best detection of relevant pathways and signature genes, highlighting it as the most suitable method for label-free proteomics.
缺失值(MVs)的存在极大地降低了无标记定量蛋白质组学数据的完整性。插补已被广泛应用于处理 MVs,选择适当的方法对于插补的准确性和可靠性至关重要。在这里,我们进行了一项比较研究,使用大规模基准数据集和免疫细胞数据集评估了七种流行插补方法的性能。模拟的 MVs 以不同的 MV 率和非随机缺失(MNAR)率组合被合并到每个数据集的完整部分中。归一化均方根误差(NRMSE)用于评估插补后蛋白质丰度和组间蛋白质比值的准确性。还使用基准数据集比较了组间真实阳性(TP)和假改变蛋白发现率(FADR)的检测。此外,通过比较免疫细胞数据集插补后细胞激活的富集途径和特征基因,评估了处理真实 MVs 的准确性。我们观察到,插补的准确性主要受 MNAR 率而非 MV 率的影响,下游分析可能会受到插补方法选择的极大影响。基于随机森林的插补方法通过实现最低的 NRMSE、高数量的 TP 和平均 FADR<5%,以及最佳的相关途径和特征基因检测,始终优于其他流行方法,这突出表明它是无标记蛋白质组学中最适合的方法。