Posada-Céspedes Susana, Seifert David, Topolsky Ivan, Jablonski Kim Philipp, Metzner Karin J, Beerenwinkel Niko
Department of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland.
SIB Swiss Institute of Bioinformatics, 4058 Basel, Switzerland.
Bioinformatics. 2021 Jul 19;37(12):1673-1680. doi: 10.1093/bioinformatics/btab015.
High-throughput sequencing technologies are used increasingly not only in viral genomics research but also in clinical surveillance and diagnostics. These technologies facilitate the assessment of the genetic diversity in intra-host virus populations, which affects transmission, virulence and pathogenesis of viral infections. However, there are two major challenges in analysing viral diversity. First, amplification and sequencing errors confound the identification of true biological variants, and second, the large data volumes represent computational limitations.
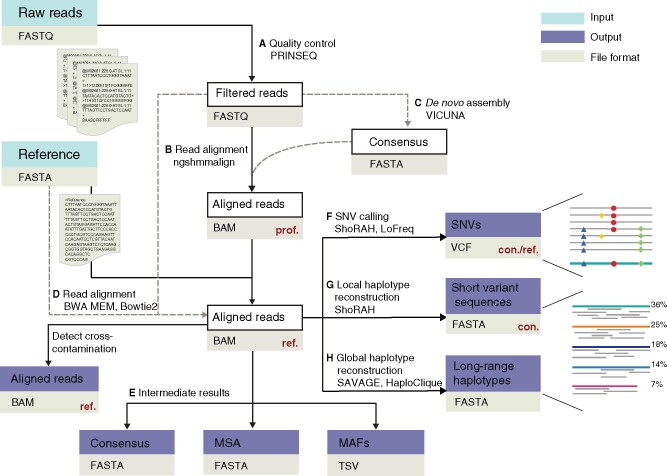
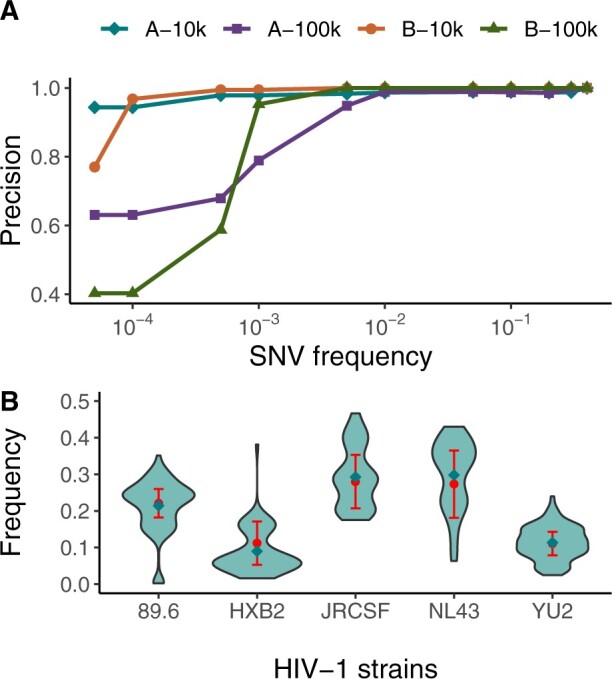
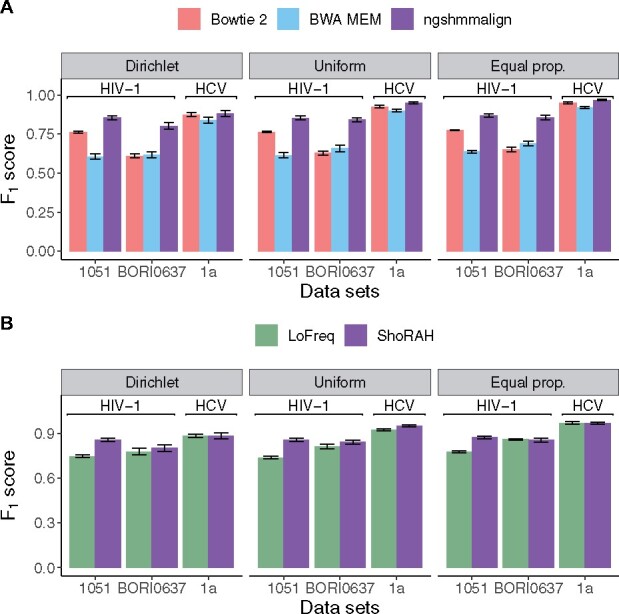
To support viral high-throughput sequencing studies, we developed V-pipe, a bioinformatics pipeline combining various state-of-the-art statistical models and computational tools for automated end-to-end analyses of raw sequencing reads. V-pipe supports quality control, read mapping and alignment, low-frequency mutation calling, and inference of viral haplotypes. For generating high-quality read alignments, we developed a novel method, called ngshmmalign, based on profile hidden Markov models and tailored to small and highly diverse viral genomes. V-pipe also includes benchmarking functionality providing a standardized environment for comparative evaluations of different pipeline configurations. We demonstrate this capability by assessing the impact of three different read aligners (Bowtie 2, BWA MEM, ngshmmalign) and two different variant callers (LoFreq, ShoRAH) on the performance of calling single-nucleotide variants in intra-host virus populations. V-pipe supports various pipeline configurations and is implemented in a modular fashion to facilitate adaptations to the continuously changing technology landscape.
V-pipe is freely available at https://github.com/cbg-ethz/V-pipe.
Supplementary data are available at Bioinformatics online.
高通量测序技术不仅越来越多地用于病毒基因组学研究,还用于临床监测和诊断。这些技术有助于评估宿主内病毒群体的遗传多样性,而这种多样性会影响病毒感染的传播、毒力和发病机制。然而,在分析病毒多样性方面存在两个主要挑战。第一,扩增和测序错误会混淆对真正生物学变异的识别;第二,大量数据带来了计算上的限制。
为了支持病毒高通量测序研究,我们开发了V-pipe,这是一个生物信息学流程,它结合了各种最先进的统计模型和计算工具,用于对原始测序读数进行自动化的端到端分析。V-pipe支持质量控制、读数映射与比对、低频突变检测以及病毒单倍型推断。为了生成高质量的读数比对结果,我们基于轮廓隐马尔可夫模型开发了一种名为ngshmmalign的新方法,该方法专门针对小型且高度多样化的病毒基因组。V-pipe还包括基准测试功能,为不同流程配置的比较评估提供标准化环境。我们通过评估三种不同的读数比对器(Bowtie 2、BWA MEM、ngshmmalign)和两种不同的变异检测工具(LoFreq、ShoRAH)对宿主内病毒群体中单核苷酸变异检测性能的影响来展示这种能力。V-pipe支持各种流程配置,并以模块化方式实现,以便于适应不断变化的技术环境。
V-pipe可在https://github.com/cbg-ethz/V-pipe上免费获取。
补充数据可在《生物信息学》在线版获取。