Eawag, Swiss Federal Institute of Aquatic Science and Technology, Dübendorf, Switzerland.
ETH Zurich, Department of Environmental Systems Science, Institute of Integrative Biology, Zurich, Switzerland.
Genome Biol Evol. 2021 Mar 1;13(3). doi: 10.1093/gbe/evab010.
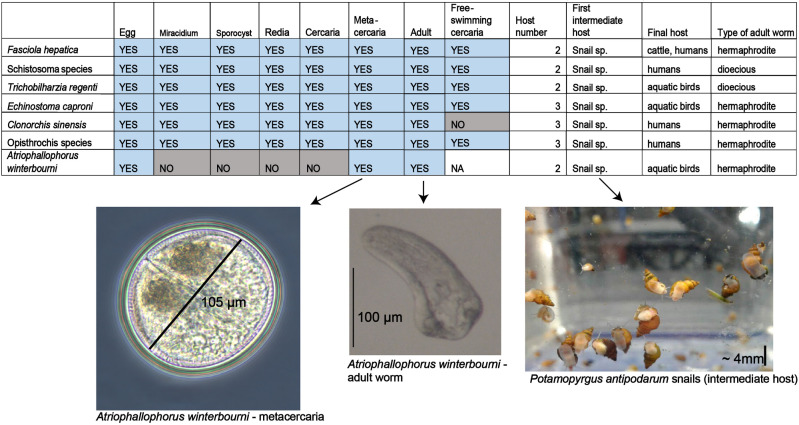
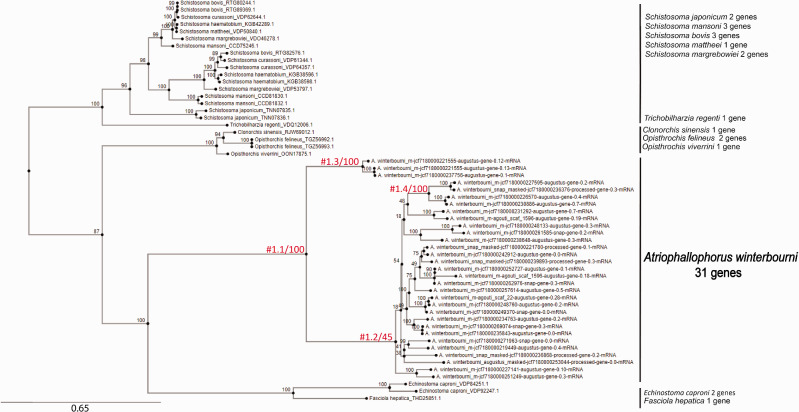
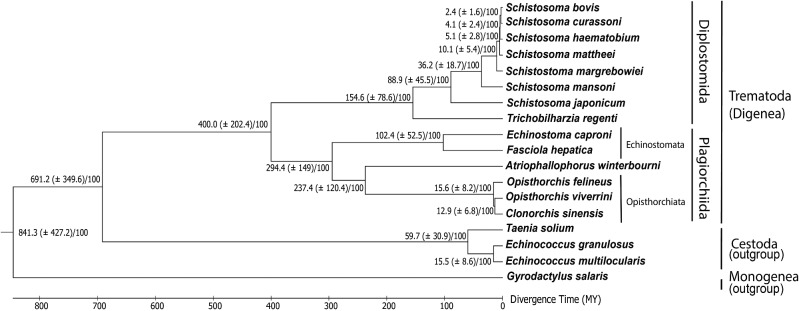
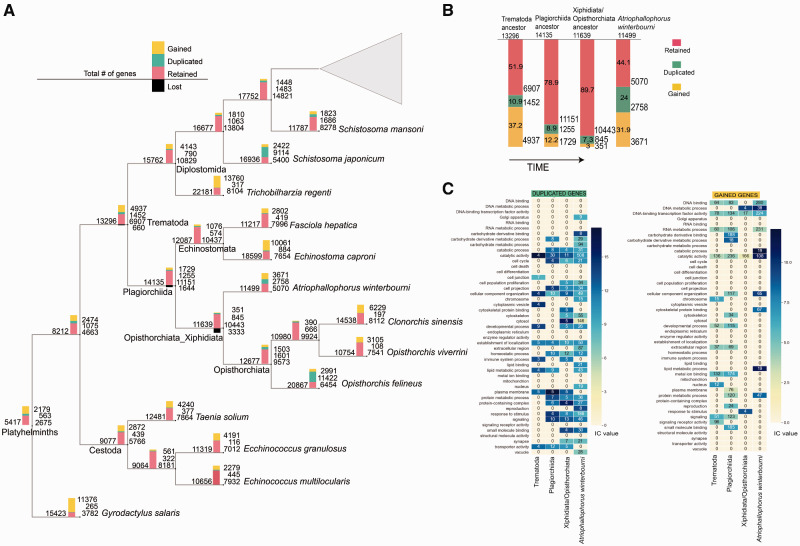
Gene duplications and novel genes have been shown to play a major role in helminth adaptation to a parasitic lifestyle because they provide the novelty necessary for adaptation to a changing environment, such as living in multiple hosts. Here we present the de novo sequenced and annotated genome of the parasitic trematode Atriophallophorus winterbourni and its comparative genomic analysis to other major parasitic trematodes. First, we reconstructed the species phylogeny, and dated the split of A. winterbourni from the Opisthorchiata suborder to approximately 237.4 Ma (±120.4 Myr). We then addressed the question of which expanded gene families and gained genes are potentially involved in adaptation to parasitism. To do this, we used hierarchical orthologous groups to reconstruct three ancestral genomes on the phylogeny leading to A. winterbourni and performed a GO (Gene Ontology) enrichment analysis of the gene composition of each ancestral genome, allowing us to characterize the subsequent genomic changes. Out of the 11,499 genes in the A. winterbourni genome, as much as 24% have arisen through duplication events since the speciation of A. winterbourni from the Opisthorchiata, and as much as 31.9% appear to be novel, that is, newly acquired. We found 13 gene families in A. winterbourni to have had more than ten genes arising through these recent duplications; all of which have functions potentially relating to host behavioral manipulation, host tissue penetration, and hiding from host immunity through antigen presentation. We identified several families with genes evolving under positive selection. Our results provide a valuable resource for future studies on the genomic basis of adaptation to parasitism and point to specific candidate genes putatively involved in antagonistic host-parasite adaptation.
基因重复和新基因被证明在寄生虫适应寄生生活方式中起着主要作用,因为它们为适应不断变化的环境(如生活在多个宿主中)提供了必要的新颖性。在这里,我们呈现了寄生吸虫 Atriophallophorus winterbourni 的从头测序和注释基因组,并对其与其他主要寄生吸虫进行了比较基因组分析。首先,我们重建了物种系统发育,并将 A. winterbourni 从 Opisthorchiata 亚目分离出来的时间追溯到大约 237.4 Ma(±120.4 Myr)。然后,我们解决了哪些扩展基因家族和获得的基因可能参与适应寄生的问题。为此,我们使用层次同源群在导致 A. winterbourni 的系统发育上重建了三个祖先基因组,并对每个祖先基因组的基因组成进行了 GO(基因本体论)富集分析,使我们能够描述随后的基因组变化。在 A. winterbourni 基因组的 11499 个基因中,多达 24%是由于 A. winterbourni 从 Opisthorchiata 分化以来的重复事件产生的,多达 31.9%似乎是新的,即新获得的。我们在 A. winterbourni 中发现了 13 个基因家族,其中有超过 10 个基因是通过这些最近的重复产生的;所有这些基因的功能都可能与宿主行为操纵、宿主组织穿透以及通过抗原呈递躲避宿主免疫有关。我们确定了几个家族的基因在进化过程中受到正选择的影响。我们的研究结果为未来研究寄生适应的基因组基础提供了有价值的资源,并指出了可能涉及拮抗宿主-寄生虫适应的特定候选基因。